
3 Silent Facility Risks That Turn FDA Inspections Into Warning Letters
April 22, 2026

3 Silent Facility Risks That Turn FDA Inspections Into Warning Letters
If the FDA walked in tomorrow, which of your “accepted” decisions would become regulatory liabilities?
A warning letter rarely starts with an inspection response. It starts earlier—when leadership approves facility, control, or validation decisions that work operationally but fail as defensible compliance positions..
That is the mistake many senior teams make just before escalation. They read warning letters as isolated failures of execution. Regulators read across systems. As the recent MD+DI coverage of Medline’s March 25, 2026 warning letter showed, CAPA failures, controlled-environment gaps, and design-verification weaknesses rarely point to one weak function. They point to an operating model already losing regulatory defensibility.
If that pattern feels uncomfortably familiar, the next step is not to review observations. It is to identify the hidden conditions that make those observations escalate.

1) The investigation system that never had a chance to find the truth
Most companies misread CAPA risk by treating it as a quality-process problem.
So they do the obvious things: revise templates, retrain investigators, add approval layers, tighten due dates, escalate review. Those actions are visible, measurable, and easy to present upward. They also often fail to fix the problem that actually matters.
Because CAPA becomes structurally weak when the operating environment does not produce clean enough evidence to support causality.
If your site runs with blurred segregation, inconsistent process signals, weak data logic, or ambiguous ownership across engineering, operations, and quality, investigations will always drift toward interpretation. QA is then asked to prove control within a system that leadership never designed to reveal the truth quickly.
That is why recurring CAPA weakness is a board-level signal, not just a quality signal. It suggests the business may be generating events faster than it can generate certainty.
The Medline warning letter makes the point clearly. FDA cited inadequate CAPA even after 221 complaints and 177 MDRs tied to syringe disconnections. The issue was not that CAPA existed. It was that corrective action did not produce control.
What should worry leadership:
If your CAPA system closes events faster than it increases certainty, the weakness is probably upstream. The facility, process architecture, or governance model may be too ambiguous to support a defensible root cause.
2) The cleanroom that looked qualified but was never truly under control
This is where many regulated manufacturers still confuse engineered environments with controlled environments.
“A cleanroom can be classified, qualified, and procedurally governed—and still be structurally weak.”
When that happens, companies usually respond the wrong way. They add more cleaning, more monitoring, more retraining, more environmental review, and more procedural restrictions.
Those are compensating controls. They are not proof that the cleanroom concept itself was robust.
In the Medline case, the FDA cited inadequate maintenance of controlled environments and noted visible particulate matter on manufacturing equipment, along with more than 100 complaints over roughly two years related to foreign matter or hair contamination, according to industry reporting. That combination matters. It signals not just a housekeeping lapse, but a control philosophy that failed to prevent contamination risk from becoming commercially and regulatorily visible.
That is why the risk of contamination is so often mispriced by leadership. It is framed as an operations issue when the real weakness sits in design logic, intervention assumptions, monitoring relevance, and control architecture.
For firms exposed to FDA, EU-GMP, and PIC/S-facing scrutiny, that matters even more. Regulators are increasingly less concerned with the cleanroom narrative and more with whether the control logic is credible under live conditions.
What should worry leadership:
If your contamination strategy depends on procedural intensity to compensate for architectural weakness, the cleanroom may be “qualified” without being reliably defensible.
3) The verification package that created the activity, not defensibility
This is where executive teams get false reassurance. Protocols, reports, retesting, and change records create comfort. But regulatory exposure disappears only when the evidence logic can withstand challenge.
Weak design verification is rarely just a protocol problem. It usually means that design intent, change impact, manufacturing representativeness, and traceability logic were never governed as a single system.
That is why some verification packages feel busy but still collapse under inspection:
- There is testing, but no proof
- There is documentation, but no rationale
- There is closure, but not defensibility
FDA’s March 25, 2026 warning letter to Medline said the firm’s rationale did not explain how testing one part could cover multiple product families and cited additional gaps requiring updates. The issue was not clerical. It was whether the manufacturer could still prove affected products met requirements after design changes.
Once verification is deemed weak, the regulator no longer evaluates a single protocol. The regulator is evaluating whether governance lost control of design intent before evidence was locked.
What should worry leadership:
If your verification logic depends on retrospective justification, the weakness is not in the report. It is in the way leadership allowed design, engineering, and validation decisions to be separate.
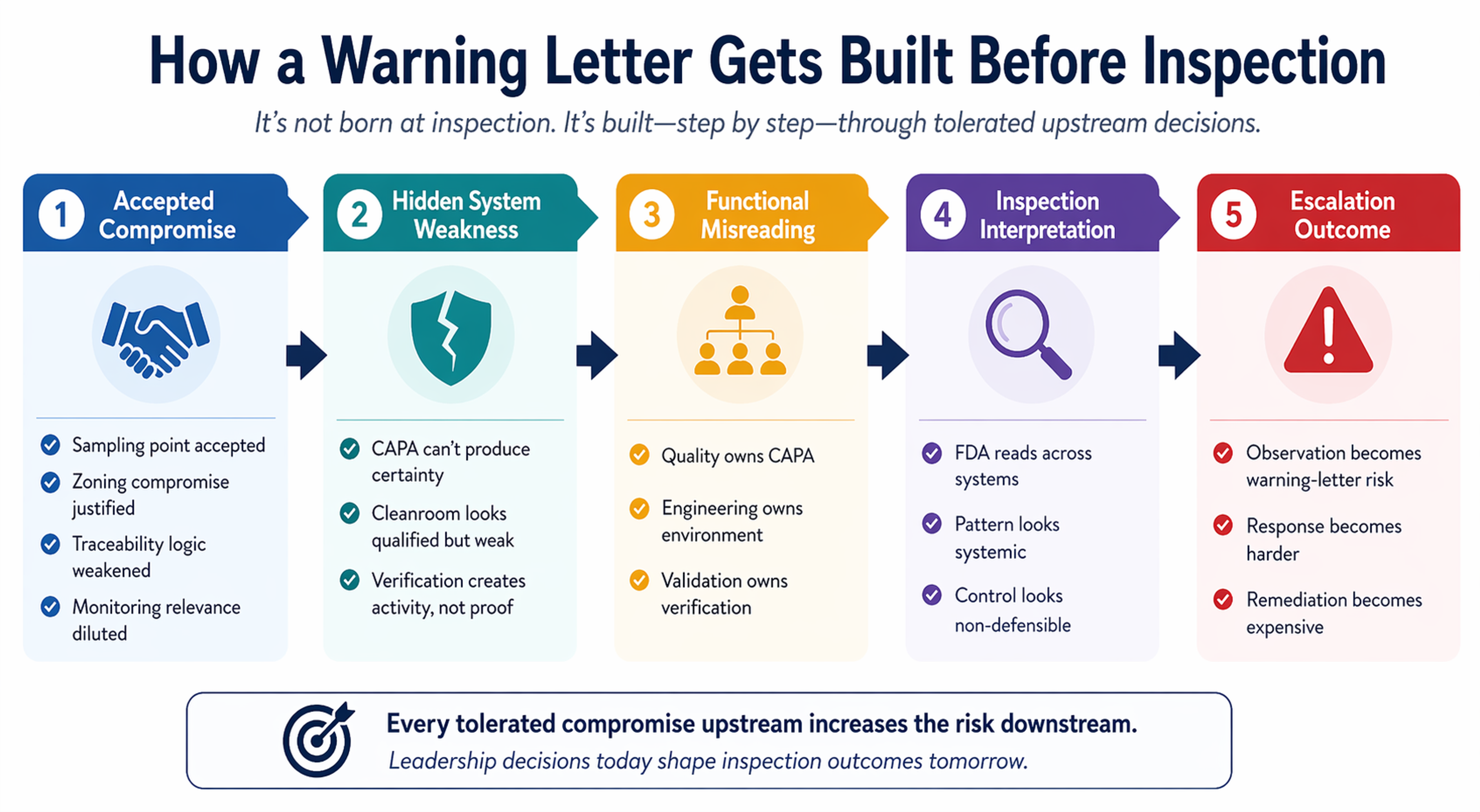
Why these three risks escalate faster than most firms expect
Senior teams often distribute these issues functionally:
- CAPA goes to Quality
- cleanroom issues go to Operations or Engineering
- verification goes to Validation, R&D, or Design Assurance
That may be administratively convenient. It is regulatorily dangerous.
Once the FDA sees weakness across investigation effectiveness, controlled-environment discipline, and verification logic, the agency is no longer looking at isolated misses. It is examining whether the company’s operating model can deliver reliable control at all.
That is why some inspections escalate unexpectedly. The site thinks it has a few issues that are containable. The regulator sees a business that keeps correcting symptoms while preserving the same underlying decision logic.
What leadership keeps mispricing before inspection
The highest-risk mistake is not noncompliance itself. It is assuming compliance can be stabilized after execution logic has already won.
Once schedule, procurement, equipment finalization, package closure, commissioning pressure, and operational convenience become the dominant project language, regulatory defensibility gets demoted. Not openly. Quietly.
A sampling point is accepted.
A zoning compromise is justified.
A representativeness argument is stretched.
A data pathway is left fragmented because the build must move.
That is how the next warning letter is often born: not in negligence, but in tolerated compromise.
And by the time inspection arrives, the organization is no longer deciding whether the design was sound. It is trying to explain why the compromise should still count as control.
Where Inotek helps leadership catch the warning-letter logic early
This is where execution-led models become dangerous. They are rewarded for momentum—closing packages, protecting timelines, preserving buildability, and minimizing disruption—not for challenging decisions that make the project easier to deliver but harder to defend later.
Inotek operates differently. It is a pure consulting organization focused on regulatory-first decisions across design and engineering consultancy, regulatory readiness, QMS consulting, and validation consulting. It is not an EPC contractor, turnkey vendor, or supply-led execution player. Its value lies in challenging weak assumptions early—before they harden into compliance exposure.
A partner tied to execution protects delivery.
A partner tied to regulatory logic protects decision quality.
And when an inspection is approaching, decision quality is usually what separates a manageable observation from a warning-letter pattern.
Test the logic—not the narrative before the next inspection
If your site is approaching FDA scrutiny, the real risk is not that one department may underperform. It is that the facility already contains one of the three silent conditions regulators keep escalating: an investigation system that cannot produce certainty, a controlled environment that appears compliant but is not fully under control, or a verification structure that generates documents without producing proof. Those are not training issues. They are strategic exposure.
If you are preparing for an inspection, upgrading a site, expanding capacity, or trying to stop repeat quality signals from escalating, bring in Inotek before execution logic hides the real problem. Inotek helps regulated manufacturers test facility strategy, contamination-control assumptions, QMS architecture, and validation logic while they are still fixable—not after regulators have already connected the dots.
To connect with our experts, visit www.inotek.co.in/contact-us.
Frequently Asked Questions:
1. Why do some facilities keep getting stuck in recurring CAPA and remediation cycles?
Because they are correcting symptoms without changing the decisions that created them, when investigation pathways, facility logic, contamination-control assumptions, and governance responsibilities were weak from the start, CAPA becomes a repetitive containment mechanism instead of a true control system.
2. Can a facility appear compliant yet remain vulnerable to regulatory escalation?
Yes. That is exactly the danger. A site can be qualified, procedurally mature, and documentation-heavy, yet still carry hidden weaknesses in segregation logic, monitoring relevance, intervention design, traceability, or validation rationale. Those weaknesses usually stay hidden until inspection pressure forces the system to prove itself.
3. What usually turns an FDA inspection into a warning-letter risk?
Not one visible lapse, but a pattern regulators can read across systems. When CAPA does not produce control, contamination-control logic looks weaker under live conditions, and verification cannot defend design intent, FDA stops seeing isolated issues and starts seeing a site-level control problem.
4. What triggers an FDA warning letter?
An FDA warning letter is typically triggered when the agency believes it has identified significant violations of federal requirements. The FDA uses warning letters to notify firms of those violations and seek prompt corrective action before moving to stronger enforcement if issues are not adequately addressed.
5. What are common FDA warning letter violations?
Common warning-letter violations include poor manufacturing practices, quality-system failures, product-claim issues, and incorrect directions for use. In pharma, biotech, and device environments, that often translates into recurring CAPA weakness, contamination-control failures, design-control or verification gaps, and broader CGMP/QMS breakdowns.
6. What are the 4 types of FDA inspections?
The FDA describes four basic inspection types: surveillance, follow-up, for-cause, and application-based inspections. Surveillance inspections assess ongoing compliance, follow-up inspections verify corrective actions after prior issues, for-cause inspections investigate specific concerns, and application-based inspections support product or facility approvals.



