
CAPA vs Design: Root Cause of Compliance Failures
April 07, 2026

CAPA vs Design: Unmasking the Real Source of Pharma Compliance Failures
How many repeat deviations, environmental excursions, or CAPA closures does it take before leadership admits the problem is no longer execution, but design?
Most organizations misdiagnose recurrence as an execution gap. At the leadership level, that assumption is expensive. In regulated environments, CAPA rarely fails due to structural issues; it fails because it is applied to problems created upstream—by engineering choices, fragmented system architecture, and weak capture of design intent.
Regulators do not reward closure velocity. They assess whether the system was designed to maintain control under stress, peak loads, human interaction, and real-world variability. When the same failures reappear, the message is clear: the control strategy lacks structural integrity. This is where the conversation must move—from CAPA efficiency to regulatory‑first design and engineering intelligence.
Inotek brings a regulatory-first lens shaped by former EU-GMP inspectors, certified quality auditors, and seasoned facility designers who have worked with leading pharmaceutical manufacturers worldwide. We engage where risk is still reversible—before flawed assumptions become recurring deviations, redesign pressure, and inspection exposure.
Why Deviations Keep Returning (Beyond CAPA)
Even well-performing sites begin to show cracks when systems are pushed beyond ideal conditions. This section explains why recurring deviations are rarely accidental and how they signal deeper instability in design and control strategy.
Design vs Corrective Action: Why Recurring Deviations Happen
Recurring deviations are not noise; they are evidence that the system behaves differently in operation than in the design assumptions. Environmental excursions, pressure reversals, integrity failures, and OOS/OOT trends are often linked by a single thread—design assumptions that break under real‑world conditions.
Most facilities are engineered for steady‑state compliance. Inspections, however, judge performance under stress. When airflow, zoning, process flow, or system integration is inherently fragile, CAPA becomes a containment loop rather than a resolution mechanism; fundamental design changes—alterations to airflow paths, zoning boundaries, or process flows—redefine the validated state and require formal change control, impact assessment, and requalification. Treating such changes as simple CAPAs understates their scope.
CAPA can temporarily stabilize outcomes, but only a robust design eliminates the risk of recurrence. Organizations that fail to separate these paths end up with persistent CAPA cycles and no structural improvement.
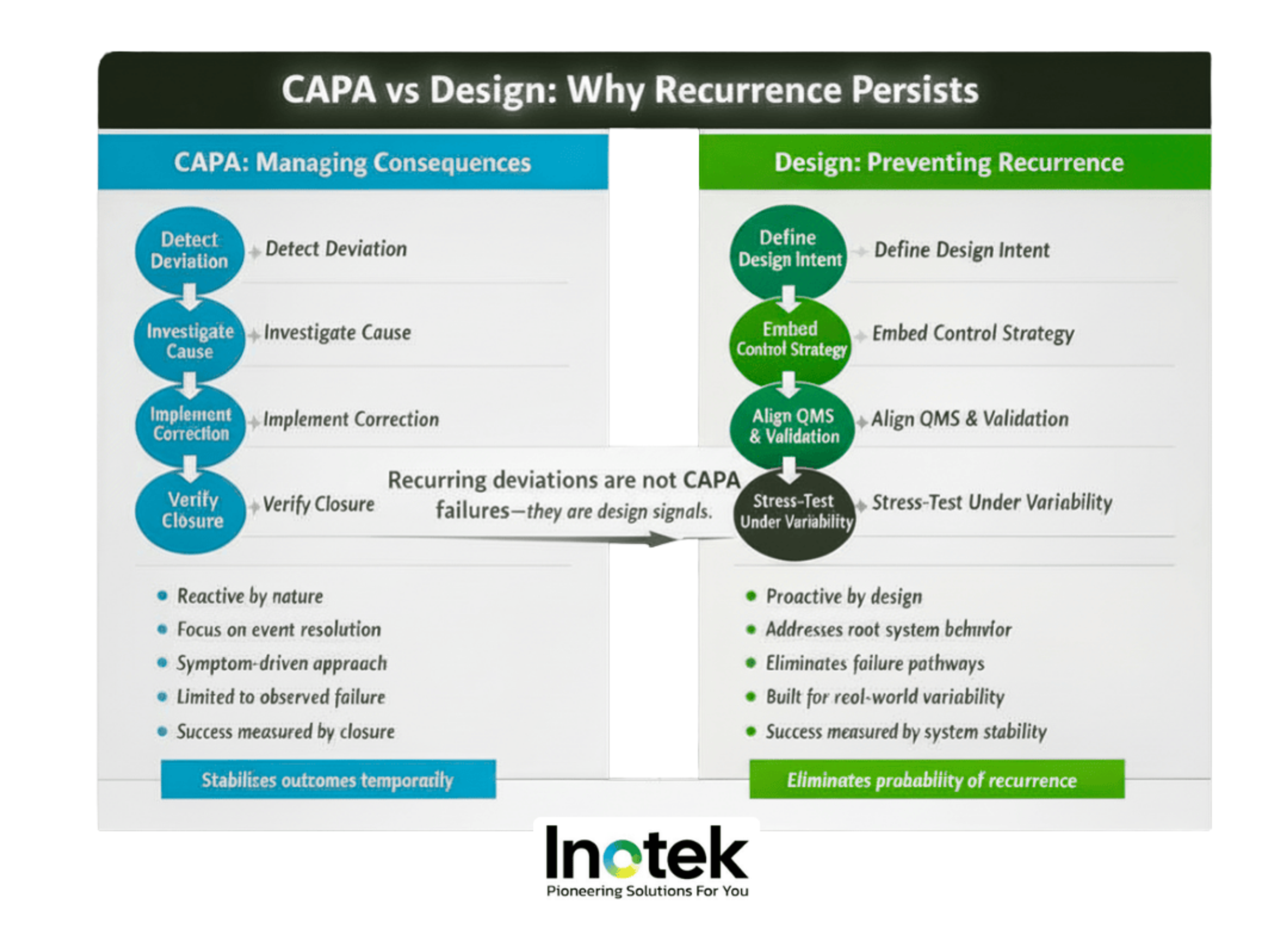
Signal vs Symptom: Interpreting FDA Deviation Patterns
Regulators interpret repeated deviations as systemic control gaps. What appear to be isolated events internally—leak tests, contamination events, or data anomalies—share a common cause: design and integration flaws that the control strategy cannot compensate for. Persistent leak‑test failures often point to vacuum or piping design limits; contamination trends trace back to airflow behavior; data issues originate from poor integration of MES, LIMS, and QMS. These are not CAPA problems. They are design problems revealing themselves through CAPA.
CAPA Matters—But It Cannot Correct Flawed Design Logic
CAPA remains a necessary part of a mature pharmaceutical quality system, but it is frequently overextended. When recurrence is driven by flawed facility logic, weak system integration, or unstable control assumptions, CAPA can contain the signal without eliminating the source.
Leadership should recognize the tipping point quickly: when the same CAPA theme survives closure, retraining, and monitoring, the issue is no longer procedural discipline. It is a structural weakness.
Why CAPA Investigations Miss Design-Level Root Causes
CAPA usually fails less in structure than in depth of investigation and system visibility. The breakdown starts when recurrence is interpreted as an execution issue, while the root cause lies upstream in design, validation logic, or system architecture.
Where leadership should look harder:
- Detection without context: Deviations are logged without mapping upstream dependencies (engineering, environment, system interaction).
- Shallow root cause analysis: Investigations stop at operator or procedural causes, avoiding design, validation, or system architecture gaps.
- Action planning under constraints: Timelines and operational pressure lead to short-term fixes rather than structural corrections.
- Verification focused on closure: Effectiveness is measured by completion, not by elimination of trend under varied conditions.
What actually proves resolution:
- No recurrence across batches and conditions
- Stability under peak load and operational variability
- Alignment between observed performance and design intent
“A closed CAPA is not proof of control. Non-recurrence is.”
When CAPA Stops Working, and Design Becomes the Only Answer
Many organizations invest heavily in CAPA yet see the same issues return. That should trigger a leadership-level realization: closure cadence and system stability are not the same thing. CAPA loops repeat because the organization is still investigating symptoms more quickly than it is confronting structural causes.
At this point, CAPA is no longer the solution. The solution is redesign—airflow reconfiguration, zoning recalibration, HVAC rebalancing—executed through change control and requalification. The distinction is decisive: containment versus elimination.
Where Design Breaks Down: The Real Drivers Behind Recurring Deviations
Recurring deviations are failed assumptions in design translated into operational signals. They expose gaps in HVAC logic, zoning, layout, CQV, and digital integration that were invisible during ideal-state qualification. This section surfaces where facility, validation, and system architecture quietly embed recurrence risk that CAPA cannot neutralize.
Hidden Design Weaknesses That CAPA Can’t Fix
Recurring deviations usually trace back to a small set of structural weaknesses. These are the design layers leadership should examine first.
1. HVAC & Environmental Control Failures: When Your Primary Control Strategy Becomes the Risk
If airflow is your control strategy, what happens when it destabilizes under load?
HVAC must sustain control across dynamic occupancy, heat loads, door cycles, and equipment variability. When it cannot, turbulence zones, pressure reversals, and recovery delays create persistent pathways for contamination.
Key failure drivers:
- Return placement creates recirculation pockets
- Insufficient recovery time after door events
- Capacity sized for average load, not peak variability
What this means for leadership: CAPA can clean up events; only engineering recalibration restores state control.
2. Cleanroom Zoning & Flow Architecture: Where Layout Decisions Become Compliance Risk
Zoning rarely fails immediately—it fails when operations scale or deviate from ideal flow.
Mixed personnel and material flows, constrained layouts, and weak segregation force workarounds. These operational compromises become contamination pathways over time.
Key risk patterns:
- Intersections between clean and dirty corridors
- Backtracking flows due to space constraints
- Staging areas are collapsing segregation logic
Implication: Once constructed, zoning errors become structural CAPA dependencies—often requiring redesign to eliminate.
3. Validation & CQV Misalignment: Passing Qualification, Failing Reality
If validation does not reflect design intent, it certifies assumptions—not performance.
When protocols are decoupled from engineering behavior, systems pass tests but fail under real operating conditions. CQV gaps leave interactions unqualified, not just components.
Key breakdowns:
- Acceptance criteria not stress-tested
- Utilities and HVAC are qualified in isolation, not in interaction
- Change impact not traced across system layers
Result: Apparent compliance with hidden instability—surfacing later as recurring deviations.
4. Human Factors & Workflow Design: Engineering ‘Human Error’ Into the System
When the system is complex, deviation is the default outcome—not the exception.
Poor ergonomics, unclear interfaces, and multi-step workflows increase cognitive load and variability in execution.
Design-induced error drivers:
- Access constraints impacting cleaning and intervention
- Interface ambiguity across digital systems
- Workflow complexity encourages shortcuts
Leadership takeaway: Training scales effort; design scales reliability. Systems must make compliant behavior the path of least resistance.
Why CAPA Loops Repeat & Root Cause Investigations Fail
Many organizations invest heavily in CAPA and still see the same issues return. That should tell leadership something important: closure speed and system stability are not the same thing.
Recurring CAPAs usually persist because investigations keep resolving what is visible, while the deeper structural cause remains untouched. The pattern typically shows up in three ways:
- Superficial investigations and symptom fixing: Teams close the immediate event but stop short of questioning design assumptions, validation gaps, or system architecture.
- Cross-functional blind spots: Engineering, QA, validation, and operations often review the issue from separate angles, leaving no one accountable for the full failure path.
- Short-term action under operational pressure: Timelines, approvals, and production pressure favor quick containment over structural correction.
Preventing Recurrence Requires Earlier Design Intervention
Preventing recurrence begins before deviation management—at the point where engineering, quality, validation, and regulatory interpretation are aligned around controllable design choices. Sites that carry a lower CAPA burden are not simply better at closure. They make fewer unstable decisions early.
1. Prevent the Same Error Earlier, Not Repeatedly
If CAPAs repeat, the system is designed to allow them.
The shift is straightforward: engineer for real operating behavior and variability, not ideal execution. That is where recurrence is prevented.
Shift in investigation lens:
- From “who failed?” → “where is error made likely?”
- From retraining → simplifying workflows and interfaces
- From SOP dependence → designing out ambiguity
Outcome: fewer CAPAs because failure probability is reduced at the source.
2. Cross-Functional Teams & Digital Tools: Why Root Causes Stay Hidden Across Functions
Recurring issues persist when no single function sees the full failure path.
Siloed views fragment investigations. The real failures sit at intersections—airflow with behavior, layout with clearance, systems with traceability.
What integrated review reveals:
- Cross-system trend visibility across engineering, QA, and validation
- Traceability between recurring events, changes, and design assumptions
- Earlier escalation of repeat signals before they harden into inspection themes
The advantage is not speed. It is an earlier recognition of structural weakness.
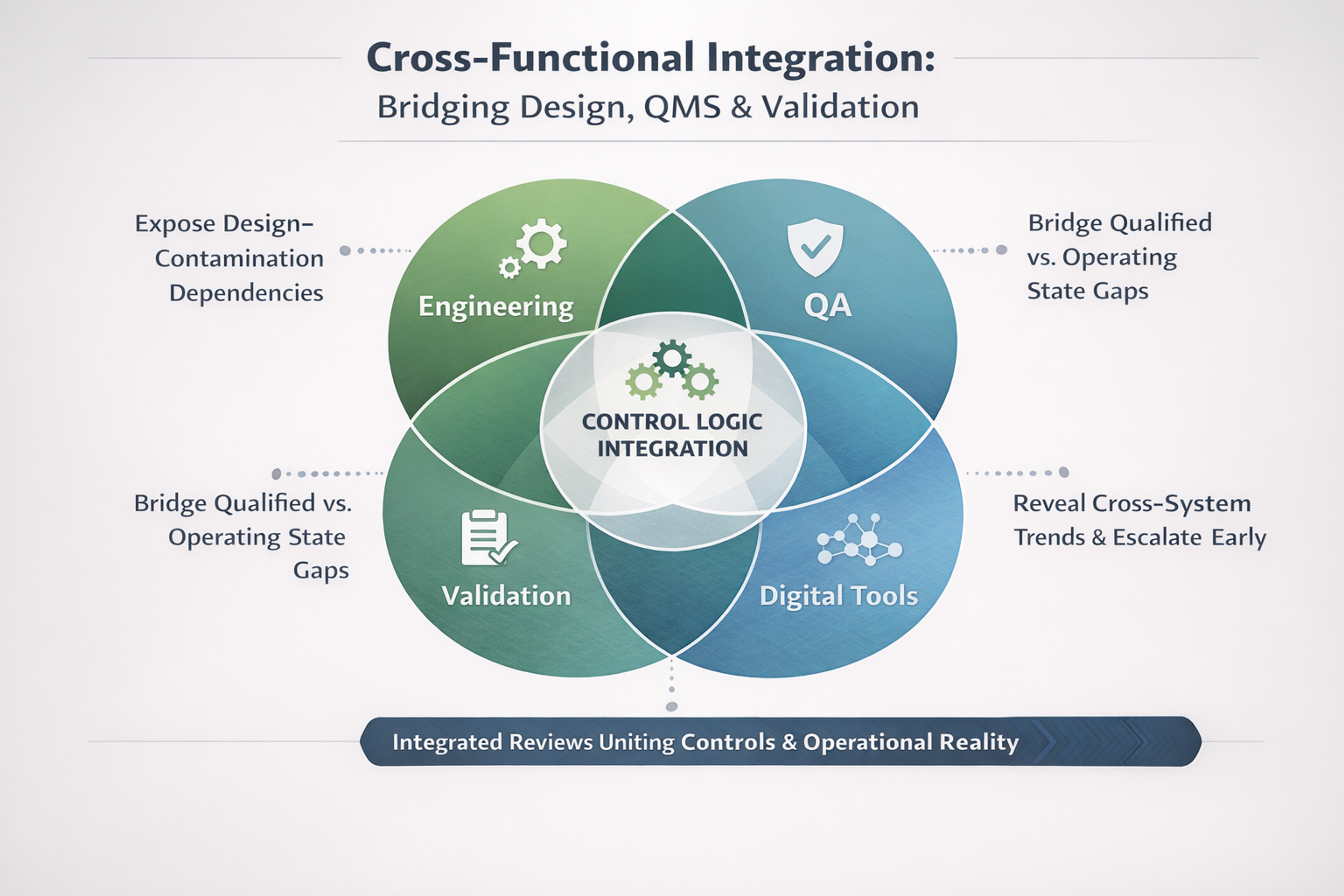
3. Aligning Engineering, QMS & Validation Strategy: Where Is Defensibility Built?
Inspection defensibility is decided when design intent becomes controlled logic.
Misalignment creates a common trap: a facility that runs, documents that pass review, and validation that qualifies—without a coherent rationale for sustained control.
What alignment requires:
- Explicit capture of design intent (URS/FDS, risk)
- QMS reflecting real engineering constraints
- Validation criteria based on operating behavior, not ideal tests
- Joint change impact across engineering, QA, and validation
Weak linkage between decision, rationale, control, and proof is where inspections start to fail.
Risk Education: Design-Linked GMP Inspection Observations
Why do observations keep pointing back to design—even when CAPAs are closed?
Inspection findings often reveal weaknesses built into the system long before the event itself became visible. When the same observation themes return across audits, regulators are not reading isolated failures. They are reading loss of control rooted in design, integration, and operating logic.
Repeated observations = design-level control gaps. If the same theme appears across audits (EM, integrity, mix-ups), regulators infer loss of state control, rather than isolated events.
What regulators are actually testing:
- Can your system maintain control under variability (not just during qualification)?
- Is your design intent traceable to QMS and validation evidence?
- Do repeated signals indicate structural weakness rather than operator error?
1. Design-Related GMP Failures: Where Early Decisions Become Audit Findings
If the layout enables risk, CAPA will keep chasing symptoms while the exposure remains built into the system.
Design-linked failures are consistent across sites and geographies because they originate from similar decision gaps.
High-frequency design observations:
- Dead legs / stagnant loops → microbial growth, cleaning validation challenges
- Zoning and segregation gaps → cross-contamination across grades
- Space and access constraints → ineffective cleaning, maintenance, and intervention
Pattern to watch: When the same category of observation appears across cycles, it is not a CAPA gap—it is a design control failure.
2. Inspection Findings & Root Cause Examples: How Design Surfaces as ‘Operational’ Failure
What looks operational in reports is often architectural in origin. That is when leadership must stop asking how the event was closed and start asking why the event was possible.
Common mappings regulators make:
- Leak-test failures → vacuum/piping design limits
- Contamination events → airflow interaction, return placement, pressure instability
- Mix-ups/line clearance issues → layout, flow sequencing, and visibility constraints
Key takeaway for leadership:
- CAPA explains the event
- Design explains why the event was possible
When those explanations diverge, inspections escalate. This is where organizations move from routine observations to repeat findings and regulatory pressure.
Reframe Your CAPA Program with Design Thinking & Risk-Based Engineering
Leadership should treat recurring CAPAs as early warnings, not administrative burdens. The real question is not how quickly the event was closed, but what its recurrence reveals about the system’s ability to hold control under real operating conditions.
Leadership lens:
- Repeat CAPAs = early inspection vulnerability
- Recurrence trends = structural, not procedural, gaps
- Stable batches under ideal conditions ≠ are controlled under stress
When the same themes persist across batches, systems, or shifts, the issue has moved beyond routine deviation management. At that point, leadership is no longer dealing with isolated events, but with an unstable design state that requires earlier, more structural intervention.
What the audit must answer:
- Where do repeat signals map to zoning, airflow, or system architecture?
- Does validation reflect operating conditions or only qualification scenarios?
- Are control limits aligned to real load, interaction, and variability?
Because the real risk is not the event itself. It is what that event proves about your system’s control integrity.
Engineer Inspection-Ready Facilities with Inotek
For organizations operating at scale, compliance cannot rely on reactive fixes. By the time recurring CAPAs or design‑linked observations appear, they are already embedded with risk, cost, and delay, as well as inspection exposure.
This is where Inotek intervenes—earlier than most, and without execution bias. Unlike EPC or turnkey providers, we operate solely as a consulting partner with no vested interest in capital execution. That independence matters: it allows regulatory science, not delivery convenience, to shape your design choices. We challenge zoning logic, facility flows, QMS assumptions, and validation strategy before weak decisions harden into recurring deviations, redesign pressure, and inspection exposure.
Our team comprises former EU‑GMP inspectors, certified quality auditors and seasoned facility designers who have worked with leading pharmaceutical manufacturers worldwide, giving you access to authority‑level expertise without the sword of capex hanging over decision‑making.
Where Inotek adds value:
– Design and engineering consultancy aligned to global GMP expectations
– Regulatory readiness across EU‑GMP, US FDA, WHO, and PIC/S
– QMS consulting embedded into operating logic, not just documentation
– Validation strategy consulting that confirms control, not assumptions
If CAPA themes are repeating, the question is not whether risk exists. It is where it is embedded, how long it has been compounding, and how much remains reversible.
Initiate a focused, regulatory-first audit with Inotek to identify where design, QMS, and validation are no longer holding up under real conditions. Once those weaknesses become visible to inspection, correction is no longer procedural. It becomes structural, slower, and far more expensive. Get in touch with us at www.inotek.co.in/contact-us.
FAQs
1. How can leadership tell when recurring CAPAs point to a design problem rather than an execution gap?
When the same issue returns after closure, retraining, and increased monitoring, the weakness usually sits deeper than execution. Repetition across batches, shifts, or systems is a strong sign that the problem is structural.
2. When does a recurring deviation require redesign rather than another CAPA?
Redesign becomes necessary when CAPA can only contain the event, not prevent its return. If airflow, zoning, layout, validation logic, or system integration are creating repeat instability, the solution must move beyond procedural correction.
3. What should a regulatory-first audit examine before weaknesses become inspection-visible?
It should test whether repeat signals map back to design assumptions, control limits, operating variability, and cross-functional gaps between engineering, QA, and validation. The goal is to identify where documented controls and actual operating behavior have begun to diverge.
4. How does Inotek differ from EPC contractors or turnkey vendors?
Inotek operates exclusively as a regulatory-first consulting partner and does not execute or supply capital projects. That independence allows facility design, QMS, and validation strategy to be shaped by regulatory defensibility rather than construction or equipment bias.



