
Publisher
India Pharma Outlook
published at
July 16, 2025
Sterile Lyophilizer Design: Inotek’s End-to-End Approach
Why do biologics lose potency before reaching patients, even with advanced cold chains?
Because traditional drying and storage methods can’t guarantee molecular stability or sterility at scale, for manufacturers handling vaccines, monoclonal antibodies (mAbs), or cell-based products, this isn't just a technical setback; it's a threat to regulatory compliance, product integrity, and commercial success.
The solution?
Sterile Lyophilization. But not just any freeze-dryer setup, one engineered with process control, aseptic assurance, and lifecycle validation in mind.
This article explores the end-to-end approach to designing, validating, and scaling sterile Lyophilizer systems. From understanding how freeze-drying preserves biologics to the regulatory-ready infrastructure needed to support high-performance operations, you’ll get:
- A breakdown of the Lyophilization process and why it matters for biologics
- Design strategies for sterility, efficiency, and compliance
- Execution workflows, from DQ to commercial handover
- Metrics that matter: throughput, PAT readiness, and qualification timelines
- Engineering insights into modular builds, SCADA/PAT integration, and contamination control
Whether you’re planning a greenfield facility, retrofitting a sterile suite, or validating a new biologic, this guide walks you through the critical decisions and systems behind effective freeze-dryer deployment.
What Is Lyophilization and Why Is It Critical in Biologics Manufacturing?
Lyophilization, also known as freeze-drying, is a sterile pharmaceutical manufacturing process used to improve the long-term stability and shelf life of temperature-sensitive biologics. It is especially beneficial for products such as vaccines, monoclonal antibodies (mAbs), enzymes, and recombinant proteins that degrade quickly in liquid form.
According to the FDA Inspection Guide on Lyophilization of Parenterals, Lyophilization offers several regulatory and operational advantages:
- Product stability: Lyophilised drugs retain bioactivity for extended periods without refrigeration.
- Sterility assurance: The process supports aseptic manufacturing with reduced contamination risk.
- Batch uniformity: Controlled process parameters ensure consistency across vials.
- Transport efficiency: The dried format reduces cold chain dependency, simplifying global logistics.
- Rapid reconstitution: Products can be reconstituted at the point of use, making them practical in clinical settings.
For facilities producing high-value biologics, Lyophilization is not only a product preservation tool, but it is a strategic necessity to meet sterility, regulatory, and operational expectations.
How Freeze-Drying Preserves Biologic Stability and Enhances Pharma Operations
Freeze drying (Lyophilization) is essential in biologics manufacturing, preserving molecular structure and potency while enabling long-term stability, sterile delivery, and simplified global logistics. The process offers both scientific and operational advantages.

How Freeze-Drying Works
Freeze-drying removes water from biologic formulations while maintaining molecular integrity through three key stages:
Freezing
- The product is cooled below the eutectic or glass transition temperature.
- Ice crystals form, separating water from active components.
- Controlled nucleation (e.g., depressurisation, ice fog) improves drying efficiency and consistency (MDPI).
Primary Drying (Sublimation)
- Vacuum removes ice without damaging the product structure.
- Heat from shelves supports phase change from solid to vapour.
- Sensors like MTM, NIR, and heat flux probes monitor sublimation to prevent collapse.
Secondary Drying (Desorption)
- Bound water is removed at higher temperatures.
- Moisture levels are reduced below 1–2%.
- Ensures long-term chemical and physical stability.
These stages prevent thermal degradation and oxidation seen in liquid or spray-dried forms.
Core Advantages in Pharma Manufacturing
Freeze drying offers more than just molecular preservation it enables robust, GMP-compliant manufacturing of biologics at scale. From extending product shelf life to reducing cold chain dependence, Lyophilization addresses critical production, regulatory, and distribution challenges in pharma facilities. Below are the key advantages.
Extended Shelf Life and Stability
Lyophilised drugs remain stable for months or years at room temperature (Cytiva). This reduces cold storage burden and transit risks, enabling:
- Stockpiling of essential biologics like vaccines and mAbs
- Fewer replenishment cycles
- Lower expiry-related wastage
- Reliable supply in remote regions
Freeze-dried products also retain potency, appearance, and sterility (PharmaGxP).
Reduced Cold Chain Dependency
Unlike liquid formulations, freeze-dried drugs don’t need constant refrigeration. As noted by MDPI, this results in:
- Lower transport costs
- Simplified global documentation
- Fewer validation checkpoints
- Easier last-mile delivery
This is critical for rapid deployment during emergencies like pandemics.
Suited for Heat-Sensitive, High-Value Biologics
Freeze drying is ideal for mAbs, proteins, peptides, and genetic vaccines. Success depends on:
- Uniform freezing and nucleation
- Controlled pressure/temperature gradients
- Minimal residual moisture (Springer)
Equipment must include:
- Cleanroom-compatible, aseptic zones
- Real-time PAT monitoring (NIR, Raman)
- CIP/SIP systems for sterility by design
Combined, these ensure regulatory compliance and product quality at scale.
Key Stages of the Freeze-Drying Process
The freeze-drying process offers clear advantages over other drying techniques, particularly for sensitive biologic formulations. Its strength lies in the DOIED model: Design, Optimisation, Implementation, Evaluation, and Documentation as defined in Springer’s review on freeze-drying process parameters:
1. Design
- Formulation and container compatibility are assessed.
- Equipment capabilities are matched to product needs (e.g., shelf cooling rates, condenser capacity).
2. Optimization
- Cycle parameters such as shelf temperature, chamber pressure, and time are optimised to balance drying time and product quality.
- PAT tools (e.g., Raman, NIR, MTM) help monitor critical quality attributes in real time.
3. Implementation
- The validated cycle is run under cGMP-compliant cleanroom conditions.
- Automated systems ensure precise control and repeatability.
4. Evaluation
- In-process controls and endpoint determinations are used to evaluate moisture content, structure, and potency.
- Visual inspection, thermocouples, and product temperature mapping are part of this step.
5. Documentation
- All parameters are recorded in accordance with validation protocols.
- IQ/OQ/PQ frameworks ensure regulatory compliance and traceability.
Compared to air drying or spray drying, Lyophilization is superior for heat-sensitive, sterile products due to its low-temperature, low-pressure environment. It protects product integrity while enabling compliance with FDA and EMA expectations for sterile manufacturing.
Sterile Lyophilizer Design: What Really Matters
Designing a sterile Lyophilizer goes beyond hardware; it’s about ensuring process consistency, aseptic integration, and long-term sterility. A key focus is nucleation control, which directly impacts drying performance, product uniformity, and compliance.
Ice Nucleation Control & Uniform Freezing
Controlled nucleation minimises batch variability by ensuring uniform ice crystal formation across vials. Without it, random nucleation leads to inconsistent crystal sizes, affecting sublimation and final product quality.
According to ScienceDirect, common techniques for uniform nucleation include:
- Depressurisation (pressure-drop method)
- Ice fog introduction
- Vacuum-induced nucleation
These methods enable synchronised ice formation, improving drying efficiency and cake uniformity. Integrating nucleation control early in design and validation ensures batch consistency, critical for sterile biologics.
Heat & Mass Transfer Engineering Using PAT Tools
Stable freeze-drying requires precise control of heat and mass transfer. Variations in temperature, pressure, or sublimation rate can cause vial collapse, hot spots, or incomplete drying.
PAT tools like NIR, Raman spectroscopy, and MTM (Manometric Temperature Measurement) play a vital role. As MDPI highlights, these tools:
- Monitor temperature and moisture in real time
- Detect issues like incomplete sublimation
- Support validation and process adjustments during scale-up
Designing for uniform shelf temperatures, cleanroom zoning, and SCADA/CIP integration enhances reproducibility, a priority for QA teams and facility planners.
CIP/SIP and Sterility Assurance by Design
CIP (Clean-in-Place) and SIP (Steam-in-Place) systems are essential for maintaining aseptic conditions throughout the Lyophilization cycle. As per Millrock Tech, effective design includes:
- 316L stainless steel chambers/shelves
- Automated steam sterilisation with pressure/temperature control
- Validated spray nozzles and drain paths
- HEPA-filtered air with controlled dew point
Designing with CIP/SIP from the outset ensures regulatory readiness and repeatable sterility, reducing audit risks and batch release delays.
End-to-End Execution: From Planning to Commercial Handover
Freeze dryer implementation isn’t complete with equipment installation alone. For regulated biopharma manufacturing, successful deployment demands a structured execution plan, starting from user requirement specifications to final performance qualification. This ensures the system delivers both sterility assurance and product consistency, in compliance with global GMP standards.
Project Planning & Design Qualification (DQ)
The Design Qualification (DQ) stage ensures that the selected Lyophilizer system meets the intended use and regulatory expectations before procurement and fabrication begin.
As outlined in the NCBI’s Lyophilization Validation Course, the key components of this phase include:
- User Requirement Specification (URS): Defines the system’s functional, regulatory, and performance needs
- Design Review: Ensures that engineering designs align with URS and GMP
- Risk Assessment: Identifies critical components and potential failure points early
- Documentation: All decisions must be traceable and compliant with ICH Q8/Q9/Q10
Early collaboration between QA, engineering, and procurement teams is essential during DQ to avoid retrofit risks and revalidation costs later.
Freezing Validation & SIP/CIP Qualification
Freeze-drying success starts with validated freezing and CIP/SIP system performance. The uniformity of ice formation impacts product stability, while validated sterilisation ensures aseptic assurance.
According to LyophilizationWorld, freezing validation includes:
- Shelf temperature mapping
- Ice nucleation reproducibility
- Product temperature monitoring across vial positions
For CIP/SIP validation, protocols must include:
- Pre and post-sterilisation bioburden testing
- Verification of clean-in-place coverage and flow paths
- Steam-in-place pressure, temperature hold times, and chamber mapping
- Cycle reproducibility over multiple runs
These validation activities are critical for maintaining both sterility assurance and batch-to-batch consistency during full-scale operation.
Lyophilization Cycle Development & PPQ Execution
Post-installation, each product requires a tailored Lyophilization cycle. This involves tuning time, temperature, and vacuum parameters to ensure optimal drying without collapse or over-drying.
As recommended by PCI Pharma Services, cycle development includes:
- Defining critical process parameters (CPPs): shelf temp, chamber pressure, hold times
- In-process monitoring using PAT tools (MTM, NIR, TDLAS)
- End-point determination using residual moisture, visual inspection, and reconstitution testing
- Execution of Performance Process Qualification (PPQ) over three consecutive commercial-scale batches to prove consistency
PPQ also documents equipment performance under load and validates the robustness of control strategies for long-term manufacturing.
Installation, Commissioning, OQ & PQ
Final validation activities ensure the Lyophilizer performs as intended under GMP manufacturing conditions. Based on Springer’s freeze dryer qualification guidance, this phase includes:
- Installation Qualification (IQ): Confirms all components are installed per specifications; includes calibration, materials of construction, and utilities
- Operational Qualification (OQ): Verifies that individual system functions (vacuum, refrigeration, shelves, automation, alarms) operate within defined limits
- Performance Qualification (PQ): Confirms the system meets performance criteria during simulated or live production runs, typically involves full load, multiple cycles, and PAT verification
This IQ/OQ/PQ framework ensures regulatory compliance and readiness for inspection. It also builds long-term confidence in system integrity, both from a quality and engineering standpoint.
Key Metrics for Evaluating Freeze Dryer Performance
To ensure consistent, compliant, and cost-effective biologic production, evaluating freeze dryer performance goes beyond initial validation. Manufacturers and project teams need to track real-time and long-term metrics that impact operational throughput, energy use, sterility assurance, and qualification timelines.
Throughput, Cycle Time, and Energy Efficiency
According to MDPI’s research on model-based freeze-drying, the most critical performance metrics include:
- Batch throughput: Defined by the number of vials processed per cycle based on chamber size and shelf loading
- Cycle time: Duration of freezing, primary, and secondary drying phases, typically 24–72 hours
- Energy efficiency: Measured by the power used per kg of water removed, factoring in refrigeration load and vacuum pump usage
Optimising these parameters helps:
- Reduce product lead time
- Lower per-unit manufacturing cost
- Increase flexibility across different product formulations
Extend equipment lifespan by reducing overuse or thermal stress
Advanced PAT tools and SCADA integration enable real-time monitoring, allowing fine-tuning of cycle parameters to boost output without compromising product integrity.
Sterility Assurance, cGMP, and Regulatory Readiness
A freeze dryer used in sterile manufacturing must meet cGMP expectations for design, materials, and documentation. As outlined by Hosokawa Micron, key criteria include:
- Sterile boundary protection: Ensuring separation between clean zones, HEPA filtration of chamber air, and aseptic loading/unloading
- Material selection: 316L stainless steel, clean welds, and pharma-grade elastomers for compatibility with SIP and CIP
- GAMP 5-compliant automation systems: Documented and validated software that controls all critical parameters
- Validation readiness: System design must support IQ/OQ/PQ processes and maintain detailed audit trails
For QA teams and compliance officers, these design choices provide the traceability and reliability needed during EMA or FDA inspections.
Design-to-Delivery Timeline Benchmarks
Project and construction teams must plan freeze dryer implementation within realistic timeframes that align with overall facility build or retrofit schedules. Based on discussions in Elsmar Forum’s OQ and project management threads, typical benchmarks are:
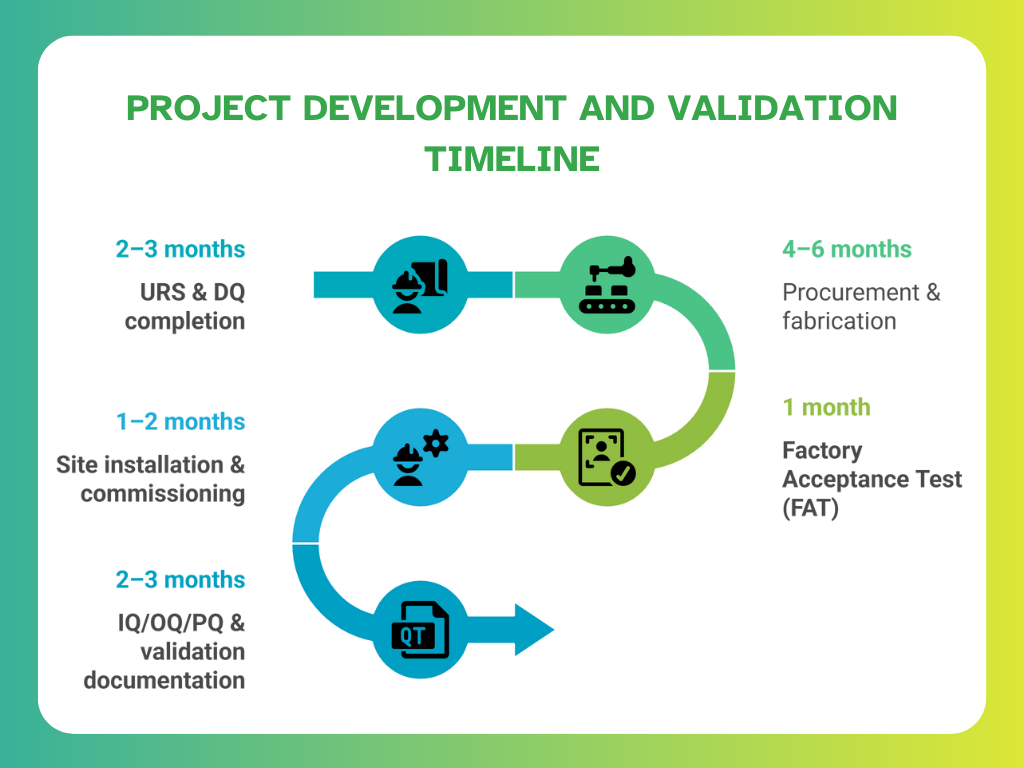
To accelerate delivery and reduce risk:
- Modular skids and isolator systems are increasingly preferred
- Pre-validated SIP/CIP systems simplify on-site qualification
- PAT-readiness reduces post-installation tuning time
Tracking progress against these milestones helps ensure accountability and minimises delays in bringing biologics to market.
Inotek’s Approach to Building High-Performance Freeze Dryer Infrastructure
Modern biologics manufacturing demands flexible, compliant, and scalable infrastructure. Freeze dryer systems must not only support sterile processing but also adapt across production scales from development batches to full commercial output. Integrating modular designs, automation readiness, and contamination control is key to delivering reliable freeze-drying performance at every level.
Modular, Scalable Builds for Lab to Commercial Scale
According to Optima Packaging’s Pharma & Biotech Solutions, modular freeze-drying systems are engineered to enable smooth scale-up across the product lifecycle. This includes:
- Pilot-scale Lyophilizers with interchangeable chamber configurations
- Commercial-scale systems are designed for isolator integration and multi-batch capabilities
- Standardised modules that simplify layout planning and reduce installation time
Each unit is designed to support SCADA systems and PAT (Process Analytical Technology) compatibility, enabling full visibility into drying parameters, shelf temperatures, and chamber pressure profiles. This modular approach reduces engineering lead time and simplifies future expansion without major retrofitting.
Automation-Ready Construction with Real-Time Monitoring
For biologics manufacturing, automation is no longer optional; it’s essential for sterility assurance, cycle repeatability, and regulatory compliance. As noted in MDPI’s paper on PAT tools in pharmaceutical processes, modern freeze dryers are integrated with:
- NIR and Raman sensors for in-line moisture and endpoint detection
- Soft sensors that estimate unmeasurable variables like residual moisture
- Remote SCADA interfaces for alarm logging, cycle control, and trend analysis
From a facility layout perspective, design teams must account for:
- Airflow patterns compliant with ISO 14644 cleanroom classes
- Pressure zoning to prevent cross-contamination between sterile and non-sterile areas
- An operator workflow that avoids disruptions to aseptic boundaries
Real-time monitoring ensures each cycle is executed within validated parameters, while automation reduces human error and simplifies batch documentation.
Containment and Cross-Contamination Control Engineering
To maintain aseptic conditions, freeze dryer infrastructure must be built around high-grade containment systems. As outlined in Optima’s Isolator & Containment Solutions, this includes:
- RABS or isolator enclosures integrated with the Lyophilizer chamber
- HEPA-filtered unidirectional airflow to eliminate particulate contamination
- Decontamination systems (e.g., VHP generators) that validate chamber sterility between campaigns
- GMP-grade material transitions and pass-throughs that prevent cross-zone exposure
For engineering and QA teams, this ensures:
- Minimised contamination risk during loading/unloading
- Reduced downtime between campaigns
- Compliance with Annex 1 and ISO cleanroom zoning principles
These design features reinforce performance reliability and long-term compliance for sterile freeze-drying operations, especially when handling high-value biologics like mAbs, peptides, and vaccine platforms.
Conclusion: Building Sterile Lyophilization Infrastructure That Performs and Complies
Sterile Lyophilization is no longer a specialised add-on; it’s a central pillar of compliant, scalable biologics manufacturing. From preserving the integrity of heat-sensitive molecules to enabling room-temperature global distribution, freeze drying offers unmatched advantages when engineered correctly.
But those advantages depend entirely on how well your Lyophilizer is designed, validated, and integrated. Every step from ice nucleation control and PAT-enabled monitoring to modular construction and CIP/SIP compliance directly impacts product quality, audit readiness, and operational throughput.
For pharma teams and project stakeholders, success lies in a unified, end-to-end approach that aligns equipment design, facility layout, and regulatory expectations from day one.
Build Future-Ready Freeze Dryer Systems with Inotek’s Expertise
The complexities of sterile lyophiliser design, from controlled nucleation and PAT integration to CIP/SIP validation and scale-up demand specialised expertise and compliance-focused execution. This is where Inotek steps in as your strategic partner.
We don’t just understand freeze-drying mechanics; we engineer regulatory-ready, high-performance freeze-dryer infrastructure into every facility we deliver, aligned with global standards including FDA, EMA, WHO-GMP, and CDSCO.
Our comprehensive approach includes:
Modular, SCADA-Integrated Design:
Customisable freeze-dryer units engineered for lab-to-commercial scale, with seamless automation and PAT compatibility built in.
Aseptic Infrastructure Planning:
Cleanroom zoning, isolator-ready layouts, and airflow design that ensure sterility by design, not just through SOPs.
Validation-Ready Execution (IQ/OQ/PQ):
Structured commissioning workflows and digital validation support that reduce delays and inspection risks.
Cycle Development and PPQ Support:
Support for product-specific Lyophilization cycles with data-driven tuning to meet regulatory endpoints.
End-to-End Project Delivery:
From URS and design qualification to FAT, installation, and commercial handover, all managed under one roof.
By partnering with Inotek, pharma manufacturers have achieved:
- 30–40% faster project qualification timelines
- Measurable reduction in validation deviations and audit gaps
- Enhanced compliance with Annex 1, ISO 14644, and GAMP 5
- Optimised layouts that improve throughput without increasing footprint
Looking Beyond Compliance
While compliance forms the foundation, successful sterile freeze-drying facilities must also address growing expectations around:
- Sustainability in HVAC and utilities
- Data integrity through real-time PAT and SCADA
- Supply chain flexibility and process scalability
At Inotek, we ensure your Lyophilization systems are not only audit-ready but engineered for long-term operational resilience and future-proof performance.
✅ Ready to Transform Your Lyophilization Facility?
Recognised among the Top 10 Pharma Turnkey Contractors & Project Consultants in 2022 & 2025, Inotek helps pharma leaders design, build, and scale sterile facilities that meet the strictest GMP and sustainability benchmarks.
📞 Connect with our experts today or visit www.inotek.co.in to schedule a consultation with Mr. Rohit Ochaney.
Whether you're planning a greenfield sterile block or upgrading your freeze-drying systems, Inotek ensures your project is compliant, efficient, and future-ready.
FAQs
What is Lyophilization in sterile manufacturing?
Lyophilization, or freeze drying, is a sterile process used to preserve sensitive biologic drugs by removing water under vacuum. It ensures long-term stability, maintains sterility, and reduces cold chain reliance, making it ideal for vaccines, mAbs, and other high-value pharmaceutical products.
How does freeze drying preserve biologics?
Freeze drying preserves biologics by freezing the formulation and removing water through sublimation. This avoids thermal degradation and oxidation, helping maintain molecular structure, bioactivity, and potency. The low-temperature, vacuum-controlled process ensures stability without compromising product integrity during storage and distribution.
What makes a Lyophilizer GMP compliant?
A GMP-compliant Lyophilizer includes aseptic design, validated CIP/SIP systems, material traceability, automation with audit trails, and robust qualification (IQ/OQ/PQ). It must meet FDA, EMA, and Annex 1 guidelines to ensure sterility, process repeatability, and data integrity throughout manufacturing and inspection.
What is CIP/SIP in freeze dryers?
CIP (Clean-in-Place) and SIP (Steam-in-Place) are automated systems in freeze dryers that ensure internal surfaces are cleaned and sterilised without disassembly. They help maintain aseptic conditions, reduce manual intervention, and ensure sterility validation for each batch in GMP environments.
How long does Lyophilizer validation take?
Lyophilizer validation typically takes 2–3 months and includes Design Qualification (DQ), Installation Qualification (IQ), Operational Qualification (OQ), and Performance Qualification (PQ). Timelines may vary based on equipment complexity, product type, and regulatory needs. Pre-validation planning can help reduce delays and rework.


