
Publisher
Inotek
published at
October 15, 2025
The Wake-Up Call: Cough Syrup Deaths and Regulatory Response
The global spotlight on India’s pharmaceutical sector has turned uncomfortably bright. Following a series of tragic child deaths linked to contaminated cough syrups in multiple countries, regulators have launched a full-scale crackdown on drug manufacturing quality. The Central Drugs Standard Control Organisation (CDSCO) and state Food and Drug Administrations (FDAs) have intensified inspection drives across the country, focusing sharply on compliance with Schedule M, the foundational GMP guideline under India’s Drugs and Cosmetics Rules.
According to recent Times of India and Reuters reports, dozens of facilities are under scrutiny for poor process validation, substandard HVAC control, and incomplete documentation trails. Several have faced suspension orders or forced halts in production. What was once seen as a bureaucratic formality is now a strategic imperative: Schedule M compliance has become a matter of survival for India’s pharma exporters and domestic suppliers alike.
This moment marks a turning point. The era of compliance-by-declaration is over. The government’s renewed enforcement is a signal that drug quality must be built into infrastructure, not inspected in paperwork.
How the Revised Schedule M Changes the Compliance Game
The Revised Schedule M 2023 notification, released in December 2023, represents the most significant GMP reform in two decades. It transforms Schedule M from a checklist into a systems-based framework, explicitly addressing facility design, data integrity, and validation lifecycle management.
For Indian manufacturers, this is more than an update; it’s a reset. Facilities that still depend on outdated layouts, manual logs, or legacy HVAC units are now at risk of structural non-compliance. The revised norms are designed to close the gap between Indian GMP and WHO-GMP alignment, preparing the sector for international scrutiny.
What’s New in the 2023–24 Schedule M Notification
The December 2023 Gazette notification redefined core expectations for premises, plant, and equipment. The updates move beyond documentation to require scientific facility design, validated cleanroom performance, and full lifecycle validation. New clauses specifically call out:
- Design Qualification (DQ), Installation Qualification (IQ), Operational Qualification (OQ), and Performance Qualification (PQ) across utilities and equipment.
- Risk management principles aligned with ICH Q9.
- Computerized System Validation (CSV) to ensure data integrity in manufacturing and QC systems.
- Cross-contamination control requires dedicated HVAC zoning and validated cleaning processes.
These inclusions align Schedule M with EU Annex 15 and PIC/S standards, making Indian plants globally benchmarkable. Yet, they also expose the weaknesses of aging infrastructure that lacks modern utilities and automation systems.
Facility Upgrade Priorities Under Schedule M
To meet the new expectations, manufacturers must look beyond procedural compliance. The path forward involves GMP infrastructure modernization, where engineering, validation, and digital systems converge to create sustainable compliance.
1. HVAC and Cleanroom Requalification
The revised norms make HVAC performance a cornerstone of compliance. Plants must now re-map airflow patterns, pressure differentials, and particulate control strategies. Air handling units (AHUs) should be requalified with proper zoning, pressure cascades, and monitoring aligned with EU GMP Class C/D environments. Risk-based requalification, especially for shared facilities, is now mandatory. Every filter integrity test, calibration record, and environmental monitoring log must form a verifiable chain of data integrity.
2. Digital Validation and Documentation Systems
Paper-based batch records, Excel logs, and standalone instruments are no longer sufficient to withstand regulatory scrutiny. Facilities should transition to validated electronic documentation systems with secure audit trails, version control, and user-based access management. This not only supports data integrity but also reduces audit stress by creating traceability between batches, deviations, and corrective actions.
3. Process Qualification and PQS Integration
The revised framework emphasizes pharmaceutical quality systems (PQS) as the backbone of continuous validation. Process Qualification (PPQ) must be integrated into a lifecycle model that ties together URS, FAT, SAT, and PQ documentation. Linking engineering and quality teams through a single digital validation platform ensures that every equipment qualification and process validation remains inspection-ready.
Global Alignment: Bridging Indian Plants to WHO-GMP Expectations
India’s pharma industry is a vital global supplier, but its GMP evolution has lagged behind its export ambitions. The new Schedule M compliance drive is designed to close that gap, bringing local plants closer to WHO-GMP and EU GMP expectations. However, aligning infrastructure and documentation practices with global norms is easier said than done.
Many mid-sized Indian facilities operate with legacy utilities, manual controls, and hybrid documentation systems typical of MSME pharma players. To help these businesses scale compliance affordably, we explore targeted strategies in our MSME Pharma & GMP Compliance Schedule M 2025 guide.
Organizations like Inotek have built expertise in translating global GMP frameworks into practical Indian execution models. Through design-stage GMP consultancy, CQV (Commissioning, Qualification, and Validation) support, and digital readiness assessments, such technical partners help manufacturers modernize without disrupting supply continuity.
Checklist: Schedule M Upgrade Readiness
A structured self-assessment can help QA/QC heads and engineering managers gauge their plant’s readiness for upcoming CDSCO audits. Here’s a quick operational checklist:
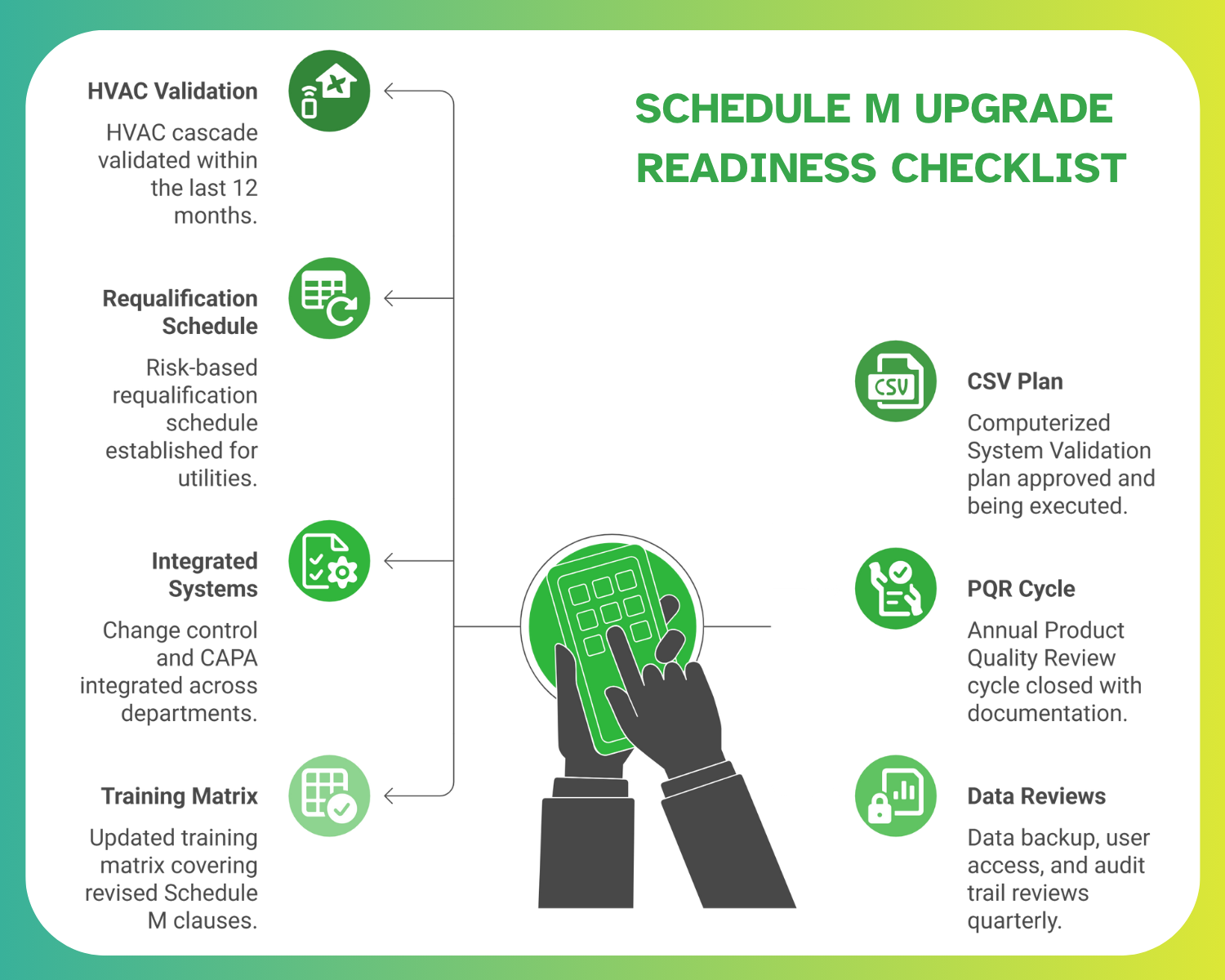
Plants that consistently meet these parameters will be well-positioned to face drug manufacturing audit readiness under both domestic and export frameworks.
The Bigger Picture: Beyond Compliance to Competitiveness
The revised Schedule M is not just a regulatory hurdle; it’s a competitiveness mandate. In an industry where buyers and regulators are increasingly risk-averse, compliance maturity directly influences business growth. Modernized facilities with validated utilities, automated records, and robust PQS are better positioned for:
- Faster regulatory approvals (domestic and international)
- Reduced product recalls and batch rejections.
- Improved energy efficiency and sustainability
- Enhanced trust with contract clients and global buyers
Moreover, Annex 1 implications for sterile manufacturing, data-driven validation (CQV 4.0), and risk-based requalification models point toward a future where digital infrastructure is integral to GMP assurance.
In this new reality, GMP infrastructure modernization is not a one-time project; it’s a continuous program that aligns regulatory, engineering, and business priorities.
Action Plan: Moving Toward Schedule M Readiness
India’s regulators have drawn a clear line: substandard GMP infrastructure and paper-driven compliance are no longer acceptable. The CDSCO’s intensified inspection drive in 2025 underscores that Schedule M compliance must now be embedded into every aspect of plant design and operation.
For pharma companies, this is the moment to act. Upgrading under Revised Schedule M 2023 is not just about passing the audit; it’s about protecting reputation, exports, and patient safety. Each delay increases the risk of non-compliance findings that can cripple production and erode market trust.
To understand how to recover from missed compliance deadlines, see our detailed roadmap in Schedule M Compliance: What to Do After the Deadline. For MSME-scale plants, our MSME Pharma & GMP Compliance Schedule M 2025 guide offers tailored best practices.
Whether you are planning a brownfield retrofit or a greenfield expansion in 2026, the key is proactive design-stage planning, digital validation readiness, and lifecycle traceability.
Inotek can help translate these regulatory expectations into actionable project blueprints, bridging the gap between GMP design, CQV execution, and audit preparedness.
👉Contact Inotek to request your Schedule M Upgrade Blueprint and ensure your facility is future-ready.
Inotek: Your Strategic Partner in Schedule M-Ready Pharma Facility Upgrades
The complexities of achieving Schedule M compliance demand specialised expertise and a deep understanding of global GMP expectations. This is where Inotek steps in as your strategic partner. We don’t just retrofit facilities for compliance; we create engineer-regulatory-ready environments that meet and exceed standards set by CDSCO, WHO, EMA, and FDA.
Our comprehensive approach includes:
1. Design-Stage GMP Consultancy: Ensuring compliant facility layouts, cleanroom zoning, and HVAC segregation from the earliest blueprint stages.
2. CQV (Commissioning, Qualification & Validation): Integrated qualification lifecycle support from URS and FAT to PQ, minimizing rework and audit delays.
3. Digital Validation & CSV Enablement: Deploying validated electronic systems that assure data integrity and streamline documentation for audits.
4. HVAC Optimisation & Requalification: Engineering efficient, validated HVAC systems with performance monitoring aligned to Annex 1 and EU GMP Class C/D standards.
5. Risk-Based Compliance Planning: Implementing periodic requalification schedules and gap assessments tailored to your facility’s operational risk profile.
By partnering with Inotek, pharma manufacturers have achieved:
- Faster commissioning and qualification timelines
- Reduced non-compliance CAPAs during WHO and CDSCO inspections
- Improved sustainability and GMP infrastructure resilience
While compliance forms the foundation, successful Schedule M upgrades must also anticipate emerging challenges such as data integrity governance, supply chain traceability, and energy-efficient facility operations. At Inotek, we ensure your projects are not just audit-ready, but engineered for long-term performance and regulatory confidence.
Recognised among the Top 10 Pharma Turnkey Contractors & Project Consultants in 2022 & 2025, Inotek helps pharma leaders design, build, and upgrade facilities that meet the strictest GMP and sustainability standards.
📞 Connect with our experts today or visit www.inotek.co.in to schedule a consultation with Mr. Rohit Ochaney.
Whether you're planning a greenfield facility or optimising an existing setup, Inotek ensures your project is compliant, resilient, and future-proof.
FAQs
What is Schedule M compliance, and why is it critical in 2025?
Schedule M outlines India’s Good Manufacturing Practices (GMP) under the Drugs and Cosmetics Rules. With the Revised Schedule M 2023, CDSCO has tightened inspection standards. Compliance is now critical for maintaining licenses, exports, and global credibility.
What are the key changes in the Revised Schedule M 2023 notification?
The December 2023 revision expands GMP requirements beyond documentation. It includes facility design standards, HVAC validation, computerized system validation (CSV), and lifecycle-based process qualification aligned with WHO-GMP and EU Annex 15.
How can pharma manufacturers prepare for CDSCO and WHO inspections?
Plants must modernize their GMP infrastructure, requalify HVAC systems, implement validated digital documentation, integrate PQS, and follow risk-based requalification. Continuous validation and data integrity are essential for audit readiness.
Why is HVAC requalification so important under Schedule M?
HVAC systems control air quality, pressure cascades, and contamination risk. The revised Schedule M mandates requalification of AHUs and cleanroom zones to meet EU GMP Class C/D standards, ensuring consistent product quality and patient safety.
What support does Inotek provide for Schedule M readiness?
Inotek offers CQV, design-stage GMP consultancy, and digital validation readiness programs. Their expertise helps pharma plants achieve Schedule M compliance, WHO-GMP alignment, and inspection-ready documentation for both CDSCO and global audits.


