
EU GMP vs US GMP: What’s the Difference and How to Stay Compliant Globally?
May 02, 2025

EU GMP vs. US GMP: A Compliance Guide for Pharma Firms and Project Managers
One regulation demands a Qualified Person. Another favors flexibility. Can your pharma facility satisfy both?
As pharmaceutical firms expand across borders, they face a significant challenge: designing and operating facilities that comply with both EU and US Good Manufacturing Practice (GMP) standards, each with its own rules for inspections, documentation, validation, and supplier qualification.
This guide cuts through the confusion. You’ll get a side-by-side breakdown of EU vs. US GMP requirements, practical insights for facility design, and proven strategies to stay compliant across regulatory zones. Whether you're expanding globally or upgrading a single site, this resource is built for teams navigating GMP from the ground up.
Introduction to Good Manufacturing Practices (GMP)
Good Manufacturing Practices (GMP) are globally recognized quality standards designed to ensure the consistent production of pharmaceutical products that meet safety and efficacy requirements. According to the WHO GMP guidelines, GMP governs areas like personnel hygiene, raw material handling, process validation, and facility maintenance.
The EudraLex Volume 4 outlines EU-specific GMP requirements, stressing the importance of quality assurance at every step of the manufacturing process. These standards directly influence how pharmaceutical facilities are designed, built, and operated- ensuring not just compliance but also production efficiency and safety.
Why GMP Matters for Pharma Facility Design
GMP is not just about documentation- it shapes critical decisions in pharmaceutical plant design. Both WHO and EU GMP stress that quality must be built into the product from the ground up. This includes facility layout, cleanroom classification, and material/personnel flow.
Per EudraLex Volume 4, pharmaceutical manufacturing sites must be designed to:
- Prevent cross-contamination
- Allow for controlled environmental conditions (e.g., temperature, humidity, airflow)
- Enable clear segregation of areas for different processes
Compliance with EU GMP guidelines is also a prerequisite for product approval in regulated markets. Regulatory bodies will assess whether the facility meets GMP expectations before approving the site for manufacturing.
For pharma project managers, early-stage alignment with GMP standards ensures that facility investments are future-proof and inspection-ready.
How GMP Affects Facility Design & Layout
WHO and EU GMP guidelines provide detailed expectations for facility infrastructure. These influence core construction decisions, particularly for high-grade cleanrooms, HVAC systems, and process zoning.
Key facility design considerations based on WHO GMP and EudraLex Volume 4 include:
- Logical material flow: Avoid backtracking or crisscrossing of raw and finished materials to reduce contamination risks.
- Personnel flow control: Segregated entry points, gowning areas, and pressure differentials help maintain cleanroom integrity.
- Cleanroom classifications (EU Grades A–D): Dictate design details like airlocks, HEPA filtration, and air change rates.
- Utilities and surfaces: Must be easy to clean and resistant to sanitizing agents, as outlined in Annex 1 of EudraLex Volume 4.
For pharma companies that design pharma manufacturing plants, these EU GMP guidelines are central to planning facility architecture, utilities, and workflow optimization. Following them ensures compliance from construction through to inspection and validation.
Understanding EU GMP: Compliance, Facility Design & Validation
EU Good Manufacturing Practices (GMP), outlined in EudraLex Volume 4, establish the baseline for ensuring pharmaceutical products are consistently produced and controlled according to quality standards. These regulations influence not only the production processes but also the architectural design, material flow, and validation protocols within pharma manufacturing facilities.
Whether designing a new plant or upgrading an existing one, aligning early with EU GMP guidelines is essential to avoid costly retrofits and regulatory delays. Compliance is mandatory for manufacturers supplying the European market.
EU GMP Regulatory Framework
The core EU GMP standards are defined in EudraLex Volume 4, which includes:
- Part I: GMP for finished medicinal products
- Part II: GMP for active substances
- Annexes: Covering specific types of production (e.g., sterile products, biologicals, computerized systems)
Regulatory oversight is provided by national competent authorities across EU member states under the coordination of the European Medicines Agency (EMA). Inspections are conducted using a risk-based approach, where facilities are audited based on the criticality of their operations, product types, and previous compliance records.
Manufacturers must ensure systems are always inspection-ready, covering everything from facility hygiene to batch documentation and data integrity.
Key Requirements for EU GMP Compliance
Achieving EU GMP compliance requires a structured approach to quality management, documentation, and validation. EudraLex Volume 4 sets expectations across several critical areas:
1. Documentation Control
All processes must be clearly documented, approved, and regularly reviewed. This includes:
- Master Batch Records
- Standard Operating Procedures (SOPs)
- Cleaning logs, calibration records, and equipment maintenance logs
- Validation protocols and deviation reports
Records must be traceable, legible, and securely stored.
2. Quality Risk Management (QRM)
A systematic, risk-based approach must be applied throughout the product lifecycle. Risk assessments are required for facility layout decisions, change control, cleaning validation, and more, ensuring that quality is built into each step.
3. Batch Control and QP Release
No product batch may be released until it is reviewed and signed off by a Qualified Person (QP). The QP ensures that every part of the manufacturing and testing process complies with GMP standards.
4. Validation and Qualification
EU GMP requires comprehensive validation activities, including:
- Design Qualification (DQ): Verifying that facility and equipment designs meet GMP requirements
- Installation Qualification (IQ): Confirming proper installation
- Operational Qualification (OQ): Ensuring systems operate as intended
- Performance Qualification (PQ): Demonstrating consistent output under real conditions
These stages ensure that facilities, systems, and processes are fit for purpose and can repeatedly deliver quality products.
Understanding US GMP: Regulations, Facility Design & Compliance Challenges
In the United States, pharmaceutical manufacturing is regulated by the Food and Drug Administration (FDA) under Current Good Manufacturing Practice (cGMP) standards. These rules ensure that drugs are consistently produced and controlled to meet quality standards suitable for their intended use.
US cGMP regulations directly affect facility layout, equipment qualification, and production workflows. Any deviation from these standards may result in warning letters, product recalls, or import alerts. For construction and project management teams, early understanding of these requirements is critical for building inspection-ready facilities.
US FDA cGMP Regulatory Framework
The legal framework for GMP in the US is based on the Code of Federal Regulations (CFR), specifically:
- 21 CFR Part 210 – Covers GMP for manufacturing, processing, packing, or holding of drugs
- 21 CFR Part 211 – Specifies GMP for finished pharmaceuticals
The FDA enforces these regulations through regular, often unannounced inspections. According to the FDA’s cGMP resource, inspections cover:
- Facility conditions and cleanliness
- Equipment maintenance and calibration
- Documentation practices
- Data integrity
- Training and qualification of personnel
The FDA emphasizes that facilities must demonstrate that processes are in a state of control, meaning they consistently yield quality product batches.
Compliance Challenges in US GMP
Unlike the EU model, which mandates specific validation and documentation formats, the US cGMP framework provides greater flexibility. While this allows companies to tailor their systems, it also creates challenges in maintaining consistency and inspection readiness.
1. Documentation Flexibility
The US model does not enforce strict formats but expects documents to be comprehensive, traceable, and available during audits. This flexibility can lead to gaps in record-keeping if SOPs and data management systems are not clearly defined and standardized.
2. Quality Risk Management (QRM)
Although risk-based thinking is encouraged, the FDA does not prescribe a single risk management framework. Firms must independently develop and justify their QRM processes. Inconsistency across departments or facilities can lead to compliance gaps.
3. Process Validation
The FDA promotes a lifecycle approach to process validation, which includes:
- Process Design
- Process Qualification
- Continued Process Verification
Unlike the EU’s structured DQ-IQ-OQ-PQ model, the US system offers more discretion in how each stage is documented and executed. This can cause confusion if not managed centrally by quality teams and project engineers.
Overall, US GMP compliance requires a well-documented but adaptable system- one that can withstand FDA scrutiny while allowing operational flexibility. For facility planners, this means designing spaces and workflows that support robust record-keeping, controlled environments, and easy access for audits and process verification.
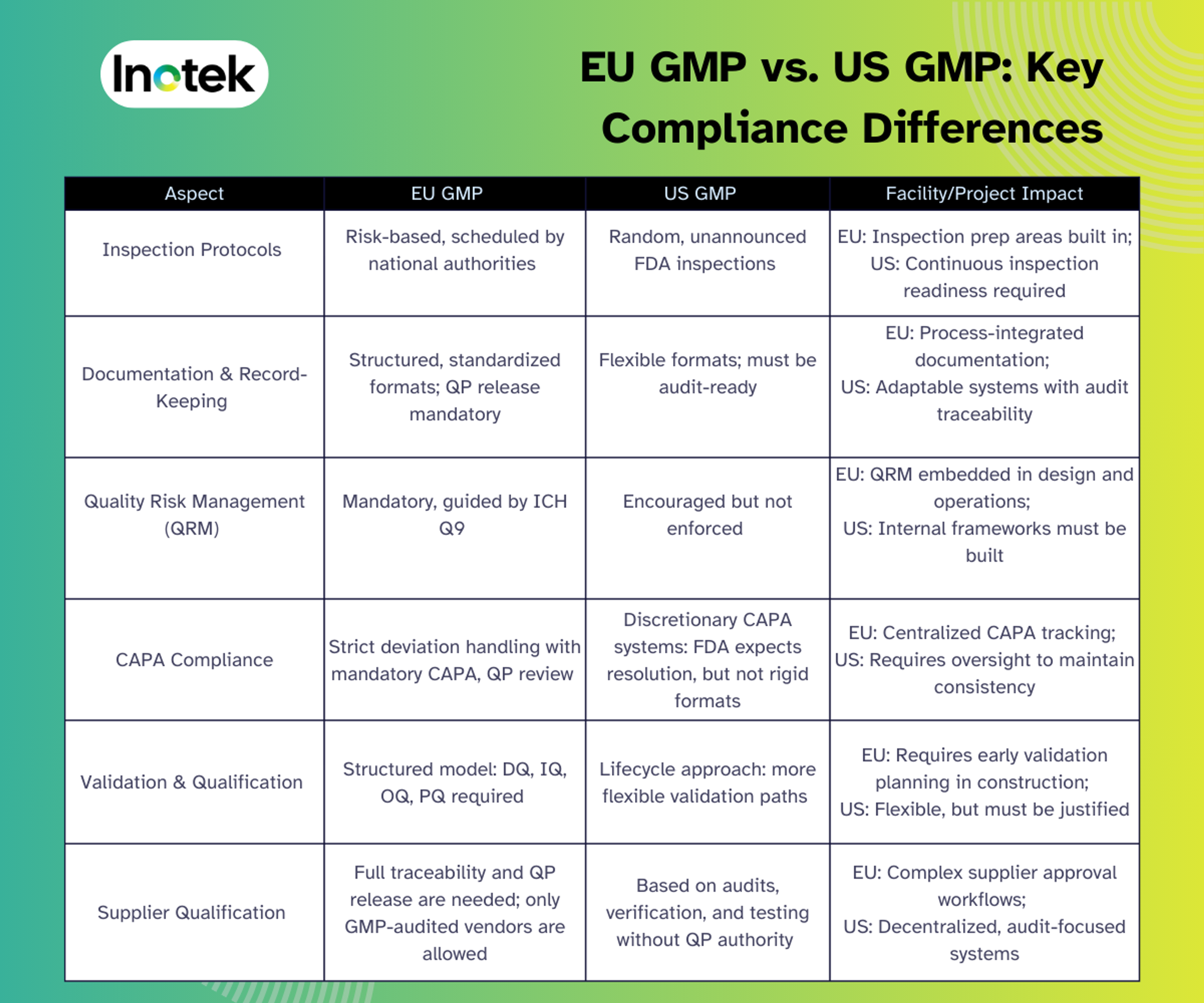
EU GMP vs. US GMP: Key Compliance Differences
While both the EU and US GMP frameworks aim to uphold product quality and patient safety, their regulatory approaches differ significantly. The table below highlights the key differences relevant to facility design, quality systems, and supplier management.
Facility Design & Construction: Adapting to Both EU & US GMP Standards
Pharma facilities serving global markets must meet both EU and US GMP standards. While both aim for product quality and safety, their design expectations differ. Early alignment helps project teams build flexible, inspection-ready spaces, minimizing compliance risks and costly rework.
Below is a comparison table to guide design decisions:
Design Considerations for Dual EU & US GMP Compliance

This dual-compliance design approach enables pharmaceutical companies to remain globally competitive, reduces inspection delays, and ensures alignment with future expansion goals in both US and EU-regulated markets.
Global Harmonization of GMP Standards: How Pharma Firms Can Stay Compliant
As pharmaceutical companies expand across regions, aligning operations with multiple GMP frameworks, such as EU GMP, US GMP, WHO GMP, and ICH guidelines, becomes essential. Although these standards share common principles, they differ in scope, enforcement, and terminology.
To stay compliant, pharma firms must build globally harmonized Quality Management Systems (QMS) that accommodate country-specific expectations while maintaining a unified operational baseline. Early integration of harmonized practices into facility design, process validation, and supplier management is key to reducing regulatory delays and inspection risks.
Challenges of Aligning with Multiple GMP Standards
Aligning operations with several regulatory standards simultaneously presents several practical difficulties:
- Terminology differences: For example, the EU requires a Qualified Person (QP) for batch release, whereas the US relies on quality units without a QP role.
- Validation models: EU GMP enforces a structured DQ/IQ/OQ/PQ model, while US GMP allows a lifecycle-based approach. WHO GMP aligns more with the EU but varies by region.
- Inspection expectations: Frequency, focus areas, and documentation depth differ significantly among regulators.
- Training requirements: Staff may need to be trained on region-specific SOPs, even for globally harmonized products.
For project and construction teams, these differences translate into more complex facility layouts, segregated documentation zones, and flexible HVAC and cleanroom systems that can meet overlapping requirements.
Regulatory Barriers to Global Pharma Expansion
Pharma firms expanding internationally face several compliance and operational hurdles:
- Country-specific GMP adaptations: Some countries have unique GMP frameworks that only partially align with ICH or WHO standards, requiring localized compliance strategies.
- Approval timelines: Regulatory review periods and inspection wait times vary widely, affecting time-to-market.
- Mutual Recognition Agreements (MRAs): While MRAs between regions like the EU and certain countries (e.g., Australia, Canada, and the US) help reduce duplicate inspections, not all countries participate. This limits the effectiveness of harmonized systems in some markets.
To navigate these barriers, companies must:
- Map out regulatory requirements early in the expansion process
- Design facilities that can be easily audited by multiple authorities
- Maintain centralized, audit-ready documentation accessible across sites
Navigating GMP Compliance for Global Pharma Expansion: Challenges & Best Practices
Expanding into new markets brings complex GMP compliance challenges. Varying regional expectations for documentation, inspections, and CAPA systems often delay approvals and increase operational risk. Companies must adopt unified frameworks that accommodate both EU and US GMP while being adaptable to local nuances. Proactive planning, strong quality systems, and cross-functional coordination are essential to streamline global compliance.
Risk Management Strategies for Global Compliance
For consistent compliance across regions, pharma companies should apply a combination of CAPA, Quality Risk Management (QRM), and process validation practices:
- CAPA: Implement a centralized CAPA system to track deviations globally. Ensure root cause analysis and resolution processes are standardized and monitored across all sites.
- QRM: Follow ICH Q9 principles to integrate risk assessments into facility design, material flow, and equipment qualification. Use QRM as the basis for change control and audit response.
- Process Validation: Align with the EU’s DQ–PQ structure while incorporating the FDA’s lifecycle validation approach. This dual approach satisfies both regulators and enhances internal process control.
Harmonizing Quality Management Systems (QMS) Across Regions
A unified QMS is essential for managing global operations efficiently. The ICH Q10 guideline provides a framework for harmonizing quality systems while allowing for regional flexibility.
Steps to harmonize QMS:
- Standardize SOPs and templates across all manufacturing sites
- Use centralized digital QMS platforms for document control, CAPA, and change management
- Apply consistent quality metrics for tracking performance, complaints, and audit readiness
- Ensure local regulatory adaptations are integrated into the global system as appendices or regional modules
This approach supports both global governance and local execution, enabling smoother inspections and fewer compliance issues.
Training & Workforce Readiness for Global GMP Compliance
Well-trained personnel are critical for maintaining compliance during global operations. To ensure workforce readiness:
- Implement global GMP training programs aligned with both EU and US regulatory expectations
- Standardize SOP onboarding across facilities to reduce variability in process execution
- Conduct mock audits and compliance drills, preparing teams for unannounced inspections
- Maintain audit logs and training records digitally for inspection readiness
Cross-functional teams- especially QA, production, and engineering- must receive role-specific GMP training. This ensures a uniform understanding of procedures, minimizes errors, and supports smoother site transitions during global rollouts.
Inotek: Your Expert Partner in EU GMP and US GMP Facility Compliance
At Inotek, we specialize in helping pharmaceutical manufacturers meet the rigorous demands of both EU GMP and US GMP standards- right from design to delivery. Our deep domain expertise in pharma facility architecture, utility planning, and quality system integration ensures that every facility we build is inspection-ready, audit-proof, and globally scalable.
Why Leading Pharma Teams Trust Inotek:
✔ Dual GMP Alignment Expertise – We design and deliver facilities that seamlessly meet both EU GMP guidelines and US FDA requirements.
✔ End-to-End Project Execution – From concept planning to commissioning, our experts manage every detail to eliminate costly rework and compliance delays.
✔ QMS-Integrated Facility Design – We embed quality systems and validation pathways directly into your facility’s infrastructure.
✔ Global Regulatory Readiness – We prepare your plant for audits across regions- whether you're pursuing EU GMP certification or expanding into US-regulated markets.
✔ Future-Ready Infrastructure – Scalable, modular, and designed for continuous inspection readiness.
As an early adopter of effective turnkey solutions in the pharmaceutical sector, Inotek delivers more than just compliance- we build pharma environments that accelerate approvals and unlock global expansion.
Talk to our pharma compliance experts today. Discover how Inotek can help make EU GMP and US GMP alignment your competitive advantage.
FAQs
What are the key differences between EU GMP guidelines and US GMP?
EU GMP guidelines emphasize structured documentation, Qualified Person (QP) release, and scheduled inspections, while US GMP allows flexible formats and unannounced FDA audits. EU GMP follows DQ-IQ-OQ-PQ validation, whereas US GMP uses a lifecycle approach. These differences affect facility design, supplier qualification, and inspection readiness.
How can a pharma company obtain EU GMP certification?
To obtain EU GMP certification, a pharma company must comply with EudraLex Volume 4, undergo a successful inspection by an EU regulatory authority, and ensure QMS, documentation, facility design, and validation processes meet EU GMP standards. Certification is granted after meeting all requirements during the formal audit.
What role does facility design play in meeting EU GMP requirements?
Facility design is critical to EU GMP compliance. It supports cleanroom classification, unidirectional material flow, and process segregation to prevent contamination. Proper zoning, HVAC design, and validation areas aligned with Annex 1 ensure pharmaceutical sites meet EU GMP requirements and pass regulatory inspections smoothly.
How does EU GMP compliance impact global pharma expansion?
EU GMP compliance strengthens global market access by demonstrating high manufacturing standards. It eases entry into regulated markets, supports Mutual Recognition Agreements (MRAs), and ensures product quality across regions. For pharma firms, aligning with EU GMP is key to successful global expansion and regulatory approvals.



