
Publisher
India Pharma Outlook
published at
August 6, 2025
AstraZeneca $50B U.S. Expansion: EPC and GMP Compliance Lessons for India
Are you prepared for the next wave of global pharma localisation?
With AstraZeneca committing $50 billion to expand U.S.-based GMP manufacturing, the stakes have shifted dramatically for pharma EPC and QA teams worldwide — especially in India. This isn’t just another industry announcement — it’s a signal that regulatory landscapes, tech transfer demands, and audit expectations are all being rewritten.
In this article, we break down:
- Why Trump’s 200% tariff threat is accelerating localisation.
- What AstraZeneca’s Virginia facility blueprint means for future-ready GMP design.
- How Indian EPCM firms must adapt for tech transfer, dual compliance, and export-ready commissioning.
- What QA and validation teams should immediately re-evaluate.
Let’s decode what this $50B bet truly means — and what you need to change before your next audit cycle.
Why U.S. Tariff Threats Are Forcing a Shift in Global Pharma Facility Design
Trump’s 200% tariff threat on imported drugs is forcing Big Pharma’s hand. President Trump has openly threatened tariffs as high as 200% on pharmaceuticals made outside the U.S. (fiercepharma.com), with an investigation underway to impose these duties. This isn’t just campaign posturing: reports suggest companies would get only a 12–18-month grace period to “get their act together” and relocate production before tariffs hit.
The U.S. Commerce Department has signalled its desire for “zero reliance” on foreign pharmaceutical inputs — as Commerce Secretary Howard Lutnick put it, Americans have been “reliant on foreign supply of key pharmaceutical products” for decades, and the new tariffs aim to address this structural weakness. In short, localization is no longer a commercial choice — it’s a regulatory mandate tied to national security concerns.
A push for domestic API and drug substance production is underway. Currently, approximately 75% of essential medicines in the U.S. are imported, with India supplying about half of the generic drugs and China accounting for 80% of their active ingredients. That dependency is now seen as a strategic risk. The Trump administration’s message is clear: “Make more of the medicines you sell in the U.S., in the U.S.”.
Major pharma players are responding in kind. AstraZeneca’s $50B U.S. bet is one of many recent localisation moves — Johnson & Johnson pledged $55B, Roche $50B, and Novartis $23B in U.S. investments in 2025 — all aimed at shifting production onshore. Industry-wide, branded drug manufacturing is being redesigned for domestic footprints in anticipation of these policies.
AstraZeneca and state officials formalize plans for a new Virginia drug substance plant as part of a $50 billion U.S. investment, July 2025.
✅ What your QA team needs to ask:
- Are our facility designs defensible under U.S. domestic-sourcing scrutiny and “America-first” audits?
- Can our cleanroom layouts, HVAC zoning, and GMP documentation hold up to both PIC/S and FDA inspections (a compliance crosswalk of EU and U.S. standards)?
Inside AstraZeneca’s $50B GMP Facility Design & Execution Strategy
AstraZeneca isn’t just spending money — it’s creating a blueprint for next-gen pharma facilities. By examining where and how that $50B is allocated, EPC teams can decode the new expectations for design and execution.
Virginia: AstraZeneca’s FDA-Compliant, Multi-Modal Manufacturing Hub
AstraZeneca’s planned factory in Virginia is a flagship project setting a global benchmark. The company will invest $4 billion in this single site, making it AstraZeneca’s largest single-site manufacturing investment ever. Once operational (targeted by 2030), the plant will produce active drug ingredients for some of AZ’s most important new therapies:
- an oral GLP-1 for weight loss
- an oral PCSK9 inhibitor for cholesterol management
- The hypertension drug baxdrostat
- and even combination therapies
These are complex metabolic molecules (such as peptides and oligonucleotides), meaning the facility must handle multi-modal manufacturing under one roof.
Crucially, the Virginia plant is designed for cross-functional GMP readiness. It isn’t a single-product factory — it will manufacture small molecules, peptides, and oligonucleotides side by side, leveraging advanced technologies like AI, automation, and data analytics to optimize production. In practice, this means the site’s utilities, cleanrooms, and quality systems must accommodate very different process needs without cross-contamination.
AstraZeneca executives emphasise that this investment is “about our pipeline” and leveraging local R&D innovation. In other words, the plant will double as a tech transfer hub for pipeline drugs — not just a static production site.
📌 Message to EPC teams: Future facilities must be modular and future-proof, able to pivot across drug modalities and scale up new processes quickly.
AstraZeneca’s Nationwide Facility Expansion: API, R&D, Cell Therapy Sites
Beyond Virginia, AstraZeneca’s expansion spans the U.S. map — effectively creating a distributed, specialty-focused network:
- R&D Hubs: Expansion of its R&D center in Gaithersburg, Maryland, and a new research center in Kendall Square, Cambridge, Massachusetts (the heart of biotech innovation). These moves cement AZ’s R&D presence in key talent clusters.
- Cell/Gene Therapy Facilities: New or expanded sites for advanced therapeutics in Rockville, Maryland, and Tarzana, California, geared toward cell and gene therapy manufacturing. These indicate dedicated spaces for viral vector production, cell processing, etc., with stringent GMP and containment controls.
- API and Specialty Manufacturing: Mount Vernon, Indiana, will see a continuous manufacturing expansion, and Coppell, Texas, will host a specialty manufacturing expansion. This suggests a focus on cutting-edge production techniques (e.g., continuous processing in Indiana) and possibly final drug product or packaging in Texas.
- Clinical Supply Chain: AstraZeneca is also upgrading its clinical trial supply network, including the addition of new sites, to ensure faster delivery of investigational medicines. This often entails high-flexibility facilities for small-batch production, packaging, and global distribution — with compliance to multiple regulators.
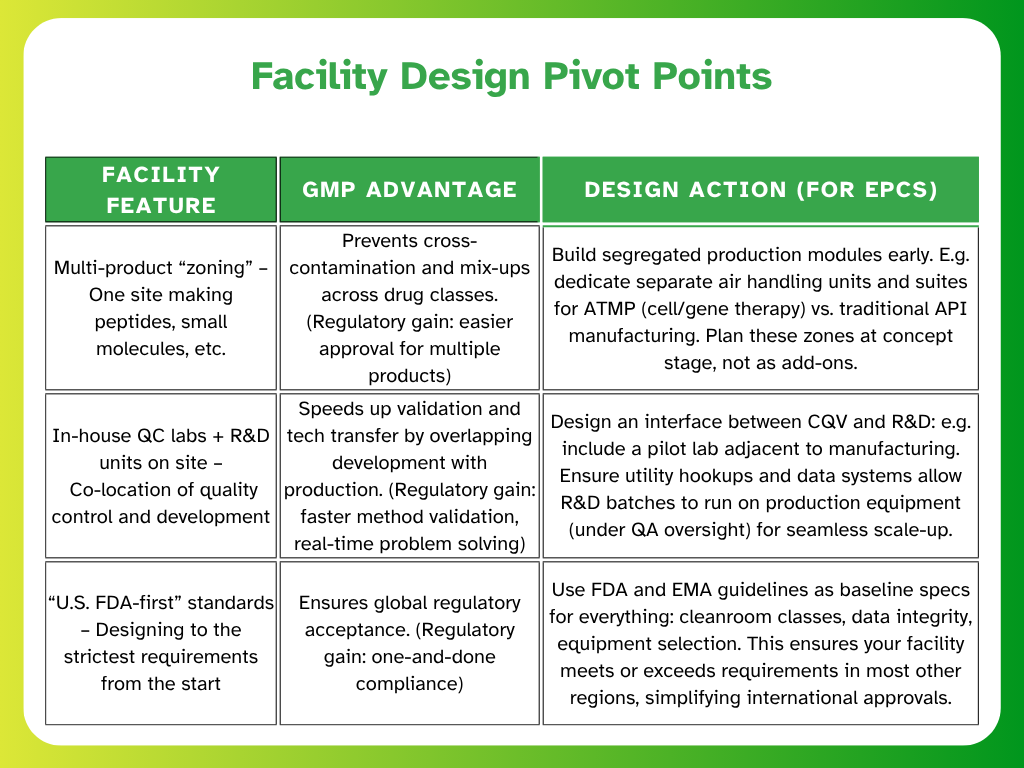
What Indian Pharma EPCM Firms Must Do Post-AstraZeneca’s U.S. Expansion
If you’re an Indian Engineering, Procurement, Construction Management (EPCM) firm working on pharma projects, AstraZeneca’s strategy is a wake-up call. The ripple effects of U.S. localisation will be felt worldwide — and “build where you sell” may soon become the norm in other regions.
Why Indian EPC Teams Must Plan for Dual GMP Compliance and Regional Localisation
Expect regulators in other markets (GCC, ASEAN, LATAM) to follow the U.S. lead and demand local production. For example, Saudi Arabia’s Vision 2030 aims to double domestic pharmaceutical manufacturing from 20% to 40% of local demand. Governments seek medicine supply security, so a facility built in India to supply the Gulf may now require a mirror facility in the Gulf in the future.
Indian EPCs must start embedding dual GMP compliance expectations from the concept stage. That means designing new plants with the flexibility to meet both local Indian CDSCO guidelines and U.S. FDA/EU standards upfront — rather than retrofitting later for export audits.
Another shift will be toward modular, quickly deployable plants with “audit-ready” commissioning packs. Essentially, regulatory documentation (URS, validation master plans, etc.) should be prepared in parallel with design and construction, so when the plant is built, it’s immediately inspection-ready for any regulator.
Firms that can deliver such turnkey compliance — not just bricks and mortar — will gain a competitive edge as pharmaceutical clients navigate rapidly changing trade rules.
Designing EPC Projects for Pharma Tech Transfer, Not Just CQV
Traditionally, EPCM teams focused on handing over a facility that passes validation (IQ/OQ/PQ). Now, the mission is broader: to build facilities that accelerate tech transfer and scale up.
Why? Global pharmaceutical majors plan to utilize their new U.S. sites as technology transfer nodes for products destined for worldwide distribution. A drug process might be developed and perfected in a U.S. plant, then transferred to satellite plants in Asia or Europe for volume production (or vice versa).
Your Commissioning, Qualification, and Validation (CQV) team must therefore enable faster process handover — e.g., by designing extra formulation suites or pilot labs for process tweaking and ensuring data integrity, so that every tweak is documented for reuse elsewhere.
The bottom line:
Don’t view validation as a one-site exercise — view it as setting up a global template that can be lifted and shifted to other locales.
This mindset shift will shorten time-to-market across regions and satisfy clients managing tech transfers alongside regulatory approvals.
GMP Execution: What AstraZeneca’s Expansion Signals for QA and Validation Teams
Quality Assurance and validation leaders on projects need to adjust their playbooks in light of these evolving trends:
1. FDA & PIC/S expectations are elevating localization to a requirement.
Regulators (FDA, EMA, PIC/S member authorities) are increasingly scrutinising supply chains. Expect questions like, “Where will you make product X for our market?”
Compliance isn’t just about the pill — it’s about where the pill is made. QA teams should prepare for facility audits that encompass supply chain provenance and traceability.
(Notably, U.S. officials have stated that “substantially all AstraZeneca pharmaceuticals sold in the U.S. will be produced in the U.S.” after these investments — a strong hint of regulatory expectation).
2. Delayed URS sign-offs will now cost you more.
In a localisation-driven build, deferring decisions is dangerous. If the User Requirements Specification (URS) doesn’t account for future regional demands (e.g, a mandate to use U.S.-sourced raw materials or add EU serialization), you risk late-stage design changes.
Lock down a globally informed URS early — or face expensive retrofits when a health authority or policy team inserts new requirements mid-project.
3. Include export-region review layers in your DQ/IQ/OQ/PQ cycles.
When qualifying equipment and systems, assume inspectors from every target market may review the records.
For instance, a new sterile line in India intended for U.S. export should undergo internal FDA-style review during DQ/IQ — catch issues before the FDA does.
Add cross-references in validation protocols to any differing requirements (e.g., EU Annex 1 for aseptic areas, or WHO GMP if relevant), so the qualification package speaks multiple regulatory languages by the time you hit PQ.
4. Beware of cross-contamination risks in multi-product facilities (see Annex 1 for focus).
As companies expand their multi-drug manufacturing sites, the complexity of preventing mix-ups increases.
The updated EU GMP Annex 1 (for sterile products) places a strong emphasis on a formal Contamination Control Strategy. QA Should enforce:
- Dedicated product zoning
- Campaign segregation
- Rigorous cleaning validation in design
…especially if a site will handle both small-molecule pills and biologics under one roof.
Overlooking these details isn’t just a compliance risk — it’s a business risk if a single contamination event can shut down a multi-billion-dollar facility.
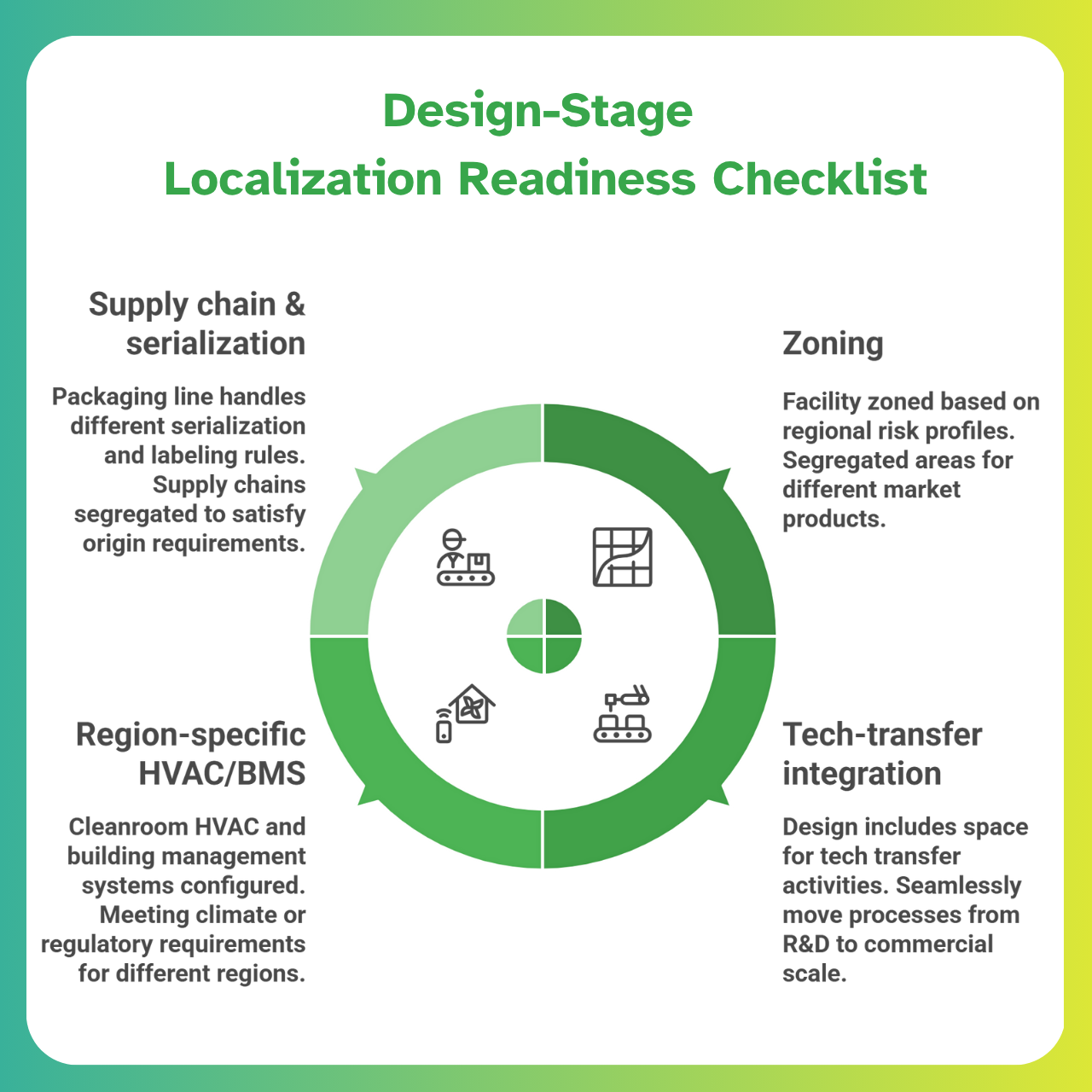
Lessons from AstraZeneca Virginia: Designing Smart, GMP-Integrated Facilities
What should EPC and QA teams glean from AstraZeneca’s Virginia mega-project?
It’s not about building the biggest plant — it’s about building the smartest, most cross-functional plant:
1. Integrated CQV strategy across drug classes
The Virginia site will produce multiple drug types (from peptides to small molecules. Such integration requires a robust CQV strategy that transcends traditional silos.
Instead of separate validation teams for each process, a unified validation master plan will oversee all, with enhanced focus on:
- Preventing cross-contamination
- Maintaining data integrity as materials move between different process streams
2.Energy and sustainability planning at the URS level
With a $4B budget, AstraZeneca is also thinking long-term. EPC teams should note that big pharma clients often bake sustainability goals (e.g., carbon reduction, energy efficiency) into project charters.
Expect requirements for:
- High-efficiency HVAC
- Green building materials
- Solvent recycling systems
…to appear in the User Requirements Specification (URS).
In Virginia, the state pitched itself on advanced manufacturing — likely including infrastructure for sustainable energy and waste treatment.
Modern pharma facilities are increasingly evaluated on their environmental GMP, so design with those KPIs in mind from the start.
3. Internal audit teams are involved in the Basis of Design stage
A key lesson from “fast-track” projects like AZ’s — negotiated in just 33 days — is that compliance cannot wait.
AstraZeneca’s own QA and internal audit specialists were likely involved from the outset to meet the aggressive timelines.
📌 Indian EPC firms should invite their clients’ QA auditors into early design reviews (Basis of Design stage).
Why this matters:
- It catches compliance issues when they’re cheapest to fix — on paper
- It aligns everyone on regulatory interpretations (FDA vs. EMA expectations) before construction begins
The result? A smoother commissioning and qualification phase with far fewer surprises.
How Inotek Interprets the $50B Shift: Global GMP Meets Indian Execution
For Indian pharma EPC teams, the writing is on the wall: we must evolve from builders into GMP-compliance integrators.
It’s no longer enough to deliver a plant on time and within budget — the plant must also:
- Meet global regulatory expectations
- Adapt to diverse local realities
The silver lining?
Our edge lies in understanding Indian execution environments plus global audit demands.
We know how to get things built in challenging conditions, and we’re fluent in cost-effective innovation.
Marrying that operational know-how with top-tier compliance expertise is our ticket to projects like AstraZeneca’s.
Start Preparing Now
CQV teams are fluent in U.S. & EU regulations
Invest in training your Commissioning, Qualification, and Validation (CQV) engineers on:
- FDA CFR expectations
- EMA/EU Annex standards
- PIC/S inspection readiness
An Indian facility targeting the U.S. supply chain should have QA documentation that reads like it was written by an FDA inspector.
Cross-train teams to enable seamless toggling between global compliance frameworks.
Fast-track risk assessments for tariff impacts
Develop a rapid-response task force to evaluate whether a client’s portfolio is exposed to trade policy shifts.
Ask questions like:
- “What if this country suddenly required local production?”
- “Can we switch to onshore execution without delays?”
Design tariff-resilient plants, with modular add-ons that can be activated quickly if production needs to shift.
This foresight positions you as a strategic partner — not just a contractor.
Pre-empt requirements — don’t wait for a perfect URS
Great EPC firms don’t just execute the User Requirements Specification (URS) — they challenge and enrich it.
If a URS from an Indian generic client doesn’t mention the U.S. FDA, that’s a red flag. Address it anyway.
Include future compliance features (e.g., space for an extra sterilizer, automated material handling, or data integrity upgrades) in design proposals.
When those “nice-to-haves” become “must-haves,” your client will remember the foresight.
Planning a 2025 expansion or export-audit-ready facility?
Learn how Inotek helps pharma teams integrate:
- Global GMP-compliant design
- Fast-track CQV execution
- Tariff-resilient infrastructure
Inotek: Your Strategic Partner in Audit-Ready, Localisation-Driven Pharma Facility Execution
The complexities of localisation-driven GMP facility execution demand specialized expertise, cross-border regulatory awareness, and audit-ready project delivery. This is where Inotek steps in as your strategic partner.
We don’t just build pharmaceutical facilities—we engineer localization resilience, dual GMP compliance, and tech-transfer agility into every project we deliver, aligned with global regulatory standards such as the FDA, EMA, and CDSCO.
Our Comprehensive Approach Includes:
- End-to-End Pharma EPC Solutions: From URS to handover, our turnkey delivery model ensures that every stage meets FDA, EU GMP, and Indian compliance requirements
- GMP Facility Design for Multi-Market Approvals: Our design approach incorporates zoning, HVAC, and documentation aligned with Annex 1, 21 CFR, and WHO TRS.
- CQV-Integrated Project Planning: We embed qualification milestones (DQ/IQ/OQ/PQ) early—so you’re audit-ready the day commissioning ends.
- Export-Audit & Localisation Strategy Support: We guide facility strategy to align with evolving “build where you sell” trends—whether for the U.S., GCC, or ASEAN regions.
By partnering with Inotek, pharma manufacturers have achieved:
- Faster facility commissioning timelines by embedding CQV early
- Reduction in cross-market regulatory gaps and CAPA risks
- Enhanced localisation alignment with sustainability and GMP goals
While compliance forms the foundation, successful pharma EPC projects today must also address:
- Environmental sustainability in HVAC and utility design
- Supply chain resilience against trade-policy shocks
- Data integrity and digital compliance across systems
At Inotek, we ensure your facility isn’t just audit-ready—but engineered for global market access and long-term operational excellence.
Recognised among the Top 10 Pharma Turnkey Contractors & Project Consultants in 2022 & 2025, Inotek helps pharma leaders design, build, and upgrade facilities that meet the strictest GMP and sustainability standards.
Connect with our experts today or visit www.inotek.co.in to schedule a consultation with Mr. Rohit Ochaney.
Whether you're planning a greenfield facility or optimising an existing setup, Inotek ensures your project is compliant, resilient, and future-proof.
FAQs
What does AstraZeneca’s $50B U.S. investment mean for EPC firms in India?
It signals a shift toward localised pharma facility design, with modular, GMP-ready, and tech-transfer-enabled infrastructure.
Indian EPC firms must now integrate global regulatory compliance from day one — not as an afterthought.
Will pharma EPC designs need to change with U.S. localisation trends?
Yes. EPC projects will increasingly need to support:
- Multi-modal manufacturing
- Audit-ready documentation
- Rapid tech transfer compatibility
…all while operating within fast-track project timelines.
How do Indian pharma EPC firms handle dual compliance for the U.S. and EU?
By embedding PIC/S, U.S. FDA, and EU Annex 1 expectations directly into:
- CQV workflows
- Facility layout
- Cleanroom zoning
This must be done at the conceptual design stage — not as a retrofit during validation or inspection prep.
What are the QA implications of multi-product facilities?
EPC designs must enable:
- Zoning and campaign segregation
- Robust cleaning validation protocols
- Formal cross-contamination control strategies
These are especially critical under EU GMP Annex 1 for sterile manufacturing environments.
How is tech transfer changing the validation strategy?
Facilities are no longer just production sites — they are now tech transfer hubs.
This requires CQV processes to:
- Ensure data integrity
- Support pilot-scale batch tweaks
- Enable cross-site process reproducibility
Validation must now serve as a template for global rollout, not just local approval.


